T2T大事件——Nature Genetics:反刍动物RT2T联盟成立
Telomere-to-Telomere (T2T) 组装技术揭示了基因组中先前“不可见”部分的新结构和功能,并允许对整个谱系中的完整基因组进行比较分析。近日,多国的科学家团队一起,在Nature Genetics上撰文介绍“反刍动物T2T联盟(RT2T)”的开放性项目合作计划,该联盟旨在生成反刍动物(Ruminantia)众多物种完整的二倍体单倍型T2T基因组,以在自然选择和家畜驯化的背景下研究反刍动物染色体和基因组演化。

一、RT2T联盟成立的必要性
反刍动物包括牛、羊和山羊等,自一万多年前被驯化以来,一直在农业中扮演着重要角色。它们将植物饲料转化为人类可消化的蛋白质,数千年来为人类提供了关键的营养来源。近年来,牦牛、水牛、野牛和一些鹿种的驯化或部分驯化,为我们提供了比较不同时间段动物驯化遗传效应的机会。此外,不同物种之间的杂交,如牛和牦牛之间的杂交,有可能为物种形成和杂交相容性提供新的见解。高质量的基因组组装不仅支持对家畜的健康、营养价值、可持续性或环境影响等性状的遗传改良,还为濒危物种的保护工作提供了宝贵资源,通过评估和维护遗传多样性来增强种群的生存能力。
由于物种界定存在争议,有蹄类哺乳动物的实际物种数量尚不清楚,部分原因是当前分类体系的修改可能导致一些物种的合并。因此,RT2T联盟的目标是通过生成高质量的T2T基因组组装,来更全面地了解这些重要动物的遗传多样性和进化历程,从而支持农业生产、遗传育种和生物多样性保护等多个领域的发展。
反刍动物种类广泛的地理分布、对多种环境的广泛适应性以及多样的染色体核型使得反刍动物成为染色体进化和物种形成理论的理想模型。比较这些物种及其近亲的完整基因组可以揭示人工选择下进化的一般原则。在这个亚目中,染色体融合/分裂以及着丝粒进化的过程随着时间的推移而反复发生,因此,完整的基因组比较可以推动对这些过程的研究。与以往基于不太全面的数据集研究反刍动物基因组的工作相比,包含着丝粒、端粒和性染色体的完整基因组组装将加深我们对这些过程的理解。
跨反刍动物谱系的T2T基因组集合提供了一个只有少数其他动物群体才能提供的机会。无论是为了农业生产还是生态保护,这些动物中的许多种类都已经是联盟成员密集进行基因组研究的对象。T2T基因组为研究超过1000万年和300多种现存物种的染色体进化提供了可能,这是使用现已过时的技术构建的参考基因组无法实现的。此外,合作的研究团队们已经是反刍动物生物学以及相关领域(如基因组组装、注释、细胞基因组学、染色体进化、基因组结构、表观遗传学和遗传多样性)的专家;他们将共同努力,一起将各自的专业知识转向揭示生物学奥秘,并可能产生对农业、健康、生态保护和生物多样性保护产生深远影响的见解。
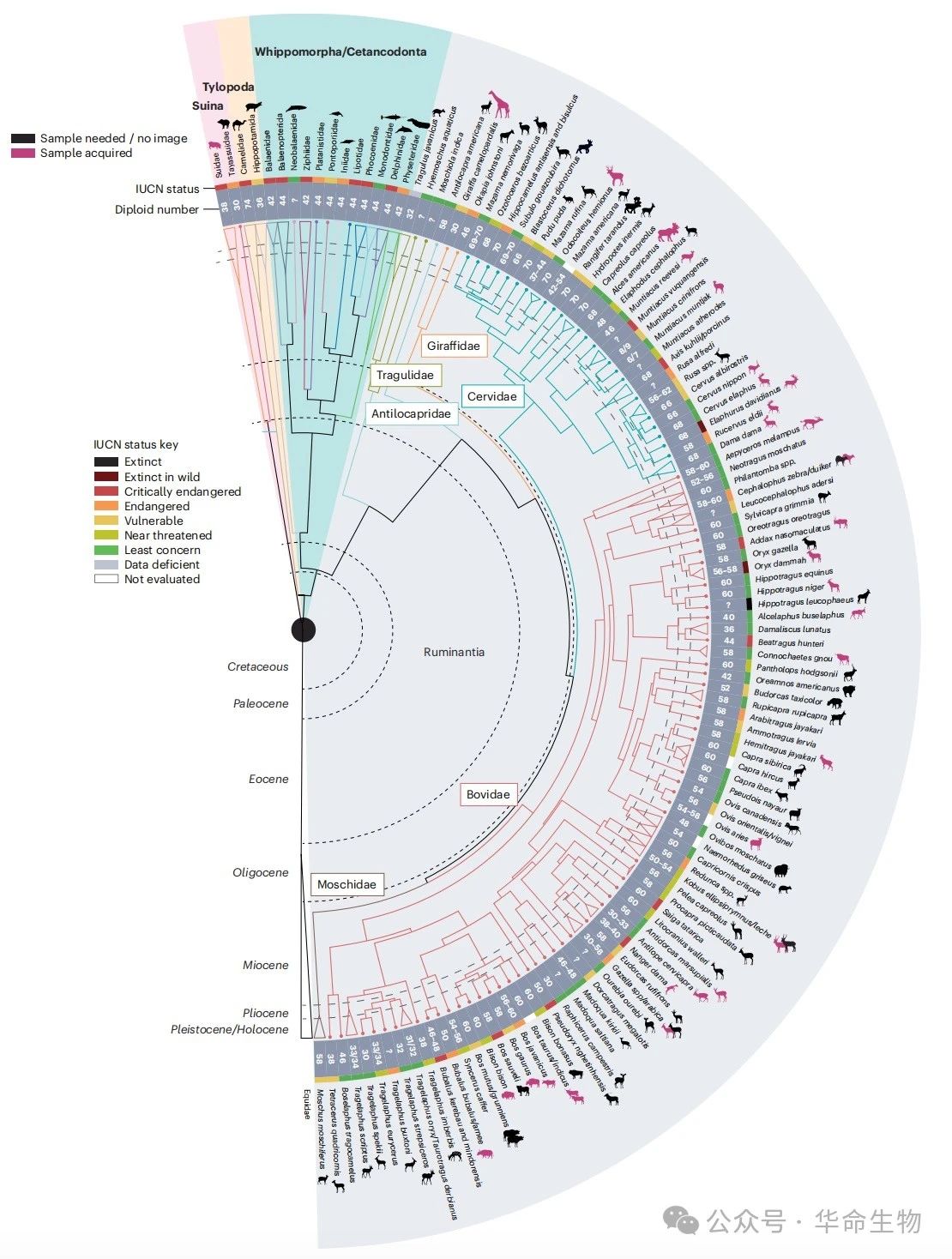
图1: RT2T计划研究的物种进化树关系图
二、反刍动物基因组学研究进展
目前,有27种反刍动物的高质量染色体水平基因组组装体达到了脊椎动物基因组项目(VGP)的N50标准(contig N50 > 1 Mb;scaffold N50 > 10 Mb),而至少有60种物种的基因组组装未达到VGP标准,这些基因组以草图形式存在于GenBank数据库中。在某些情况下,GenBank中列出的参考基因组的连续性相较于数据库中的其他组装基因组来说质量更差。例如,对于牦牛,其推荐的参考基因组的contig N50仅为22.8 kb,但存在其他三个连续性更高的牦牛基因组,其中一个的contig N50高达72.3 Mb。目前在GenBank或欧洲核苷酸档案数据库中,尚未有反刍动物的T2T基因组收录。
大部分反刍动物都比灵长类动物拥有更多的端着丝粒或亚端着丝粒染色体。例如,与人类拥有的5条端着丝粒染色体相比,牛有29条端着丝粒和亚端着丝粒常染色体,而绵羊有23条端着丝粒和亚端着丝粒常染色体以及3条亚中着丝粒常染色体。物种间固定的染色体重排可能是物种进化的重要驱动力,因为它有助于形成物种特异的基因调控模式、基因组组织、活性重复序列元件和重组模式。反刍动物在哺乳动物中拥有最广泛的染色体组成范围,其染色体数目从哺乳动物中最少的印度黇鹿的2n=6或7条,到许多鹿类物种的2n=70条不等。许多反刍动物种类以其广泛的染色体重排(分裂、融合、易位和着丝粒移动)、多种性染色体组成(例如,XX/XYY)以及潜在的由减数分裂驱动的B染色体的存在而著称,这在几种小鹿上均有发现。
三、反刍动物的T2T基因组生成
RT2T项目计划遵循HT2T(Human Telomere-to-Telomere)联盟当前的指导方针,这些方针已在人类和非人灵长类动物上进行了测试。该计划通过生成以下数据来实现:
1. 高精度的Pacific Biosciences HiFi长读长测序数据,长度在18-20kb范围内,覆盖深度为50×(每个单倍型25×)。
2. “超长”Oxford Nanopore Technologies(ONT)读长数据,长度超过100kb,覆盖深度同样为50×(每个单倍型25×)。
3. 50×的Hi-C或Omni-C短读长数据,或Pore-C长读长数据。
值得注意的是,随着ONT数据长度和准确性的提高,测序方案可以持续进行调整。目前方法需要相对大量的高质量组织、血液样本或细胞系,特别是对于ONT超长测序。为了代表两条性染色体,建议优先选择雄性个体。如果能够获取亲本样本,则可以通过对父母本进行测序来解析单倍型,从而识别出亲本特异性标记,并根据亲本来源分配组装数据。如果无法同时收集到两个亲本样本,则可以使用Hi-C数据来解析单倍型,最终目标是每个个体生成两个完整无间隙的单倍型基因组,并且在每个染色体末端都具有端粒序列(即T2T)。
RT2T计划将每种物种的组装草图提交给GenomeArk数据库(https://genomeark.github.io/)。这些组装基因组在发布后,将供任何对该物种基因组感兴趣的研究人员自由使用。RT2T将与NCBI合作,加强注释工作,以识别着丝粒并描述重复序列的类别和分布。因此,联盟鼓励对这些研究内容感兴趣的科学家们积极参与该项目。
每个组装基因组的注释工作将借助多个组织(如果有的话)的全长转录组序列来支持完成。此外,还将包括公共数据库中的数据,以进一步支持基因注释和识别在现有RNA序列数据中未观察到的转录本。在此过程中,将广泛关注与繁殖、发育、泌乳、反刍动物消化、先天性和适应性免疫、组蛋白修饰、印记基因、节段重复和逆转录基因内的基因以及异染色质区域内的基因相关的基因。同时,将重点强调可转座元件的注释,通过全面的鉴定来识别可转座元件、内源性逆转录病毒和β逆转录病毒。
这些注释工作将为研究人员提供一个更完整、更准确的基因组资源,以便他们更深入地了解这些物种的生物学特性和遗传机制。通过整合多种数据源和采用先进的计算方法,RT2T联盟期望能够生成高质量的基因组注释,为未来的基因组研究和应用提供有力支持。
四、拟成立的工作组和工作内容
RT2T目标是利用反刍动物内的比较分析,来识别染色体演化的机制,以及选择和环境压力对生物性状、基因组序列、染色质组织和结构的影响。RT2T拟成立多个工作组(Working Groups,简称WGs),每个工作组将专注于特定领域的详细分析,同时整合跨组的数据(如表1所示)。联盟计划完成大部分的物种基因组,对这些T2T基因组进行比较分析,并邀请其他研究人员加入联盟,而不是单独发表组装基因组的部分内容。不过组装基因组发布后,联盟鼓励大家立即进行这些物种的深入研究。
三维(3D)基因组工作组的目标是比较反刍动物物种内和物种间不同组织中的基因组结构。该工作组将进行染色质构象分析(如Hi-C、Pore-C和/或Micro-C),这些数据用于分析定义基因调控区室(A/B compartments)、拓扑结构域(TADs)和用于比较分析的loops。之前一项使用Hi-C技术的食肉动物物种3D基因组研究显示跨三个家族(自其最后共同祖先以来已分离5400万年)的整条染色体水平上的广泛保守性。另外,包括鸡、猪和山羊在内的家畜物种肝脏样本中的TADs和区室也显示出高度一致性。然而,关于反刍动物物种内和物种间染色体的3D构象的文献很少,而A/B室、TADs和loops的组织特异性和发育阶段特异性使比较研究变得复杂,这些分析将提供额外信息,以预测基因组变异(包括结构变异和序列多态性)对表型和适应性的影响。RT2T基因组组装的一个重要贡献是使物种间和种群内结构变异的综合分析成为可能,因为它们对3D基因组结构有重要影响,很可能影响基因组功能。在跨越后生动物系统(如人类、小鼠、果蝇、酵母)的模式物种中,最近的研究表明,拓扑结构域(TAD)边界定义了进化上保守的基因表达模式,并且响应选择的物种特异性重排往往富集在TAD边界上。因此,染色体进化工作组(Chromosome Evolution Working Group)将进行染色体和着丝粒的结构与进化的比较分析,并利用全染色体范围的三维基因组测序来探究自反刍亚目(Ruminantia)和偶蹄目(Artiodactyla)最近共同祖先以来的基因组进化。
染色体进化工作组(Chromosome Evolution Working Group)的主要目标是推导出一个祖先核型,定义反刍动物进化的断点,并从任何特定谱系特征中区分出哺乳动物中染色体结构和进化的普遍机制。这一目标的实现将需要与细胞基因组学工作组(Cytogenomics Working Group, WG)的紧密合作,后者致力于解析那些已有或可建立细胞系的物种中存在的模糊核型。荧光原位杂交(Fluorescence In Situ Hybridization, FISH)技术将被用来验证特定DNA序列在染色体上的位置,这对于那些难以组装的区域(如卫星重复序列和rDNA基因阵列)特别有用。例如,rDNA阵列中组装的间隙几乎都是相同拷贝组成的串联重复序列,可以通过高分辨率荧光原位杂交技术来解决。通过这种方法,染色体进化工作组能够更准确地重建祖先核型,识别出进化过程中发生的染色体断裂和重排事件,并探讨这些事件如何影响基因表达、遗传多样性和物种适应性。同时,与细胞基因组学工作组的紧密合作将有助于解决细胞遗传学研究中遇到的模糊问题,提高核型分析的准确性和可靠性。染色体进化工作组的研究工作不仅深化了我们对哺乳动物染色体结构和进化基本规律的理解,还为生物进化、遗传疾病、物种保护等多个领域的研究提供了宝贵的参考和依据。
注释工作组(Annotation Working Group, WG)将为那些在之前测序和组装技术所组装的参考基因组中未组装出来空白区域提供新的基因和元件注释。HT2T项目的一个成果是在新组装和校正的基因组部分中识别出以前未发现的基因,其中大多数可能为非编码RNA。然而,对于特别复杂的长非编码RNA类别,关于其在不同物种中的结构组织、功能和进化的知识仍然有限。注释工作组将利用项目中生成的数据和公共转录组数据集来注释非编码RNA以及蛋白质编码基因。通过反刍动物之间的比较研究以及基因扩张或收缩与基因组生物学其他方面的相关性研究,将为我们提供关于非编码RNA在基因组功能和进化中作用的新见解。另外还将进行编码基因家族的收缩和扩张的比较分析,并识别出处于正向选择下的基因。通过转录本丰度与顺式调控元件多态性的相关性分析,将识别出表达数量性状基因座(eQTLs),这些基因座可以阐明功能生物多样性的原理及其对进化发育、选择和适应性反应的影响。
免疫基因组学工作组(Immunogenomics Working Group, WG)专注于研究表达的适应性免疫基因,其中一些基因位于生殖系(germline)位点并通过体细胞基因组重组进行编码。反刍动物的免疫系统揭示了生殖相关区域重复适应性免疫位点的独特特征,这些位点会经历体细胞重排,但由于无法解析这些重复区域,因此一直难以研究。例如,牛能产生“超长”抗体,这可能补偿了与其他哺乳动物相比牛基因组中编码抗体的片段数量减少的情况。超长抗体在应对牛类疾病中可能起着关键作用,并可能对人类免疫缺陷病毒具有潜在的治疗应用。然而,并非所有反刍动物都能产生这些超长抗体,这使得反刍动物谱系成为研究免疫系统进化的一个非常好的模型。此外,研究反刍动物免疫系统的基因组学对于确保重要经济和濒危物种的生存以及控制可传播给人类的人畜共患疾病也至关重要。该工作组将注释编码抗体和T细胞库的生殖系基因,使用表达库测序数据(AIRR-seq)确定其表达数量性状基因座(eQTL)特征,并进行比较,以揭示与环境、病原体暴露和驯化相关的反刍动物适应性免疫系统的物种特异性适应。这种比较分析还将使我们能够研究这些所谓超长抗体的进化起源。
比较甲基化组(表观遗传)工作组将利用牛津纳米孔技术和HiFi测序读数都能识别碱基修饰这一特点,每种反刍动物物种的完整T2T基因组组装都将产生一个伴随的T2T甲基化组,从而可以研究5mC在多大程度上影响基因表达、基因组调控和基因组稳定性。首先,将在整个基因组中,包括在以前未解决的基因组区域(如rDNA阵列和着丝粒区域)中,对5mC的修饰模式进行解析。随后,对不同反刍动物之间的比较表观遗传学分析将揭示驯化对甲基化模式和基因表达的影响,这与在鱼类经过一代驯化后观察到的现象,以及在狗和灰狼之间的比较中观察到的现象相似。最后,对反刍动物中基因组甲基化模式的研究可能有助于农业基因组学了解标记辅助选择非加性性状的潜力。
变异发现和群体测序工作组的目标是确定T2T水平组装对基于短读长和长读长的变异识别和基因分型的影响。借助非常准确的长读长测序技术,T2T组装可以解决在之前组装方法中被压缩的重复序列区域。由于这些压缩区域而导致的假阳性变异体的数量将减少,相反,更完整的组装将允许在基因组中以前未解析的区域中识别变异。这些新组装区域的高准确性还将消除许多由于组装中的单核苷酸和插入缺失(indel)错误而导致的假阳性变异检测。农业上重要的反刍动物已成为继人类1000基因组计划之后多个项目的研究对象,这些项目使用了中等密度或高密度SNP芯片,以及最近的全基因组NGS序列数据。此外,这些资源已被用于建立与表型的关联。对于拥有足够群体数据的物种,变异工作组打算制作标准化资源,包括变异位置和群体等位基因频率,从而实现从以前的参考基因组到新生成的T2T基因组的过渡。最后,获得濒危反刍动物种群水平的全基因组散弹枪测序数据,将增进大家对适应性的全基因组变异分布的理解,通过对纯合基因片段的数量和分布的分析来了解物种近交情况,以及了解可能对物种适应性产生负面影响的有害变异。这些信息可用于为种群提供繁殖建议,制定种群间迁移的种群管理计划,以及其他旨在确保濒危物种长期可持续性的保护行动。此外,该工作组的比较基因组学方面将提供有关分类学和进化优势的有价值见解,区分密切相关的物种,并促进了功能性和因果性变异的识别,这在其他情况下是不可能实现的。这是物种保护不可或缺的信息,对家畜动物的基因组选择也有用处。
结语
RT2T有着宏伟的目标:为尽可能多的反刍动物和偶蹄目中的其他物种组装并注释完整的T2T基因组,以推动染色体结构和基因组进化的比较分析。对反刍动物T2T基因组的分析将提供前所未有的染色体和基因进化的分辨率,包括基因组中重复序列富集且往往具有物种特异性的区域。所获得的T2T参考基因组将有助于实现更广泛的VGP和EBP项目的目标,同时还有可能进行这些项目当前组装标准下无法进行的分析和研究。RT2T的初步成果已在GenomeArk数据库中发布,包括部分家畜完整的X和Y染色体的组装结果。
RT2T非常欢迎国际社会的参与,并鼓励感兴趣的研究人员与联盟联系。这是基因组学领域的一个激动人心的时刻,科学家们希望这样的大型开放合作能够推动整个领域的发展。
参考文献
"The Ruminant Telomere-to-Telomere (RT2T) Consortium" Nature Genetics;https://doi.org/10.1038/s41588-024-01835-2
华命生物产品服务一览