

NG重磅综述丨泛基因组在生物多样性研究的应用
完整的遗传变异数据集是生物多样性基因组学研究的关键。长读长测序技术的快速发展,使组装高质量单倍型解析的参考基因组成为可能。然而,即使是单个个体完整的T2T基因组,因为无法充分代表一个种群或物种内的遗传多样性,仍可能会使下游分析产生偏差。通过将来自同一种群、物种或属的多个高质量基因组序列信息整合到单个参考基因组上,这些基因组比对集合组装而成的泛基因组能够克服代表性偏差问题。
近日,来自美国和意大利等多国的科学家团队,在著名期刊Nature Genetics上撰文发表了名为《Pangenome graphs and their applications in biodiversity genomics》的综述文章,文章既回顾了现有的用于构建泛基因组图谱和可视化处理的工具及数据结构,同时结合实际案例,探讨了泛基因组在整个生命之树的生物多样性与保护基因组学中的重要应用。

一、泛基因组变异图谱
作者主要聚焦由组装序列全基因组比对构建而成的泛基因组图谱。变异图谱通过存储完整的基因组序列及其变异情况,更全面地呈现动植物基因组所有信息。线性基因组通过图谱的明确路径存储,而序列变异由 “气泡”或“绳结”的子图谱表示。人类泛基因组参考联盟(HPRC)生成了首个人类泛基因组变异图谱。一些模式生物(鸡等)和非模式物种(家燕、家朱雀等)的泛基因组图谱也已组装完成。一些具有重要经济价值的物种,如番茄、葡萄和牛等的超级泛基因组图谱也正在构建之中。
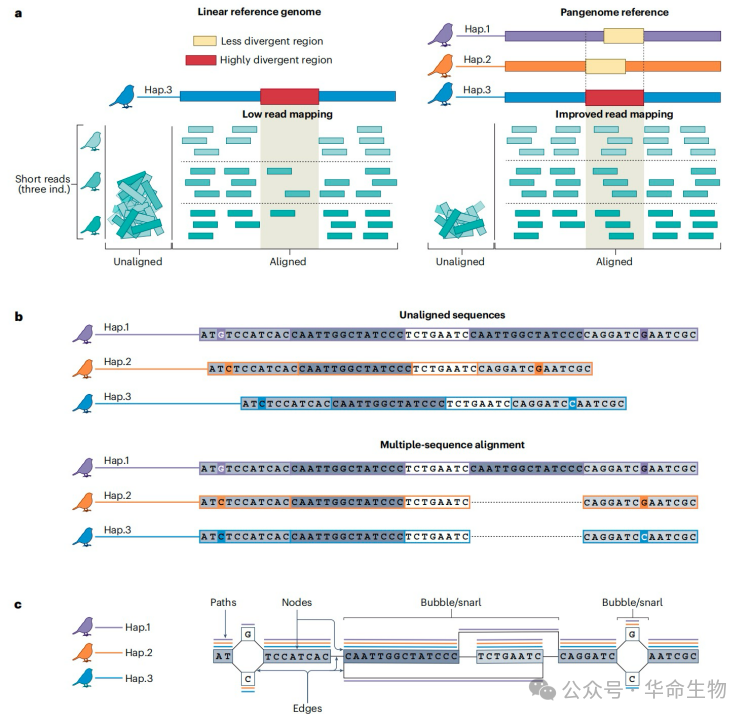
图1:泛基因组图谱
二、泛基因组采样和测序
为了最大程度获得物种的遗传多样性,合适的采样和测序策略对于构建泛基因组图谱尤为重要。
1. 合适的采样策略
采样策略对于生物多样性泛基因组研究的成功至关重要。采样应最大限度地涵盖自然种群内的基因组和生物地理多样性,理想情况是在平衡性别代表性的同时,对整个地理范围进行采样。理想样本量是在分析中逐步添加一定数量的代表性基因组,直至捕获到完整的基因集,且再添加个体也无法发现新基因。泛基因组图谱有助于明确非核心基因组区域的功能和适应性作用,以及它们在不同地理种群和亚种间的差异。这些非核心基因组区域为种群分化和物种形成提供热点区域。研究在经历杂交的种群和物种中,这些区域受基因渗入的影响程度,有助于深入了解基因流动态和物种形成过程 。
2. 基因组测序策略
对于选定个体的测序,作者更加推荐通过将 PacBio HiFi 、ONT超长和HiC数据相结合,生成完整的端粒到端粒的单倍型基因组(T2T)。T2T基因组还能更好地呈现难以测序和组装的区域,如着丝粒、可变数目串联重复序列以及其他复杂重复序列。高度重复区域也可能是调控复杂行为表型(如迁徙行为)的基础 。
低质量基因组构建的泛基因组,会因复杂区域的不完整性而限制对遗传多样性的研究。作者建议,泛基因组图谱应至少包含一个T2T水平的高质量组装基因组作为图谱构建的主干,为下游分析提供一个可靠的坐标。
三、泛基因组图谱构建
1. 泛基因组构建流程
人类泛基因组参考联盟(HPRC)开发了两条主要的泛基因组图谱构建流程:Minigraph-Cactus(MC)和PanGenome Graph Builder(PGGB)。
MC将minigraph用作图谱构建工具。用户选定的参考基因组被用作初始主干,随后逐步添加来自其他基因组的结构变异。所有组装结果会使用类似minimap2的算法回比到图谱上,为每条参考染色体生成碱基层面的比对。MC采用了无参考比对工具Progressive Cactus的改良版本,将这些比对结果整合为包含各种大小变异的碱基层面泛基因组图谱,然后将染色体图谱合并,通过折叠冗余序列来降低路径复杂度。除了选定的参考基因组,用户还可以指定其他带有坐标的组装结果,这些结果在下游分析中可作为参考 。
与MC不同,PGGB 不使用初始参考基因组,而是利用wfmash进行全基因组两两比对。seqwish作为序列到图谱的比对工具,它从全基因组两两比对开始,生成一个完整的泛基因组图谱,能够呈现所有类型和大小的变异。随后,该图谱会经过平滑和归一化的后处理步骤。在PGGB生成的泛基因组图谱中,图谱内包含的每个基因组都可作为下游分析的参考。
泛基因组图谱可以使用变异图谱工具包(vg)与转录本注释相结合,vg 是一种用于变异图谱构建、处理和分析的软件,可将其转化为剪接感知图谱,其中通过节点(外显子)和边(剪接连接点)的路径代表mRNA转录本的结构。通过将一组单倍型特异性转录本投影到一组已知单倍型上,构建泛转录组也是可行的 。
泛基因组图谱的大小取决于相应物种的基因组大小,但因为整合了其他个体的辅助序列所以会比单个参考基因组更大,同时也受到个体数量、多样性以及构建流程的影响。MC图谱的大小相对接近物种的基因组大小。相比之下,PGGB图谱的大小可能会显著超过基因组大小以及MC图谱。PGGB图谱较大的原因在于它能够捕捉高度分化的卫星区域、着丝粒区域和异染色质区域,而 MC图谱将这些区域排除在外。PGGB还有将复杂区域(如拷贝数多态性位点)压缩为单拷贝的倾向,从而在图谱中产生环,增加了图谱的复杂性。鉴于 PGGB图谱更大且更复杂,与MC图谱相比,它需要更多的计算资源。最近发布了PGGB流程的Nextflow实现版本,以提高集群的可扩展性。不过也有项目表明,PGGB可能会导致对序列变异性的高估。
鉴于存在这些差异,谨慎选择泛基因组图谱构建流程至关重要。总的来说,MC推荐应用于重测序项目作为参考,而当重点关注特定区域时,PGGB就更适合选择。总体而言,泛基因组图谱面临概念和计算方面的挑战,目前其构建、存储和分析所需的资源比线性基因组要多得多。但这些限制因素正在逐步被解决。
2. 提高泛基因组信息的可获取性
泛基因组图谱蕴含着广泛而复杂的生物信息,包括个体间的基因组关系和多样性。其错综复杂的结构产生了海量数据,在浏览和解读时颇具挑战。为改善下游分析软件对图谱的可访问性,进而便于从泛基因组图谱中提取生物信息,目前开发了泛基因组图谱多种处理工具包。主要包括排序、索引、修剪和子采样等。排序可优化图谱节点的顺序,路径索引能更快速地访问图谱的特定区域,修剪可以进一步加快计算速度,可以通过修剪复杂或不可靠的区域,或对子样本用户定义的坐标,来简化图谱拓扑结构。当计算资源不足以覆盖整个图谱,或想要梳理特定感兴趣区域的复杂性时,子采样也非常重要。
目前主要有两个用于处理泛基因组图谱的软件包:vg 和泛基因组分析工具包ODGI。vg依赖于.vg格式,是首个可扩展到千兆碱基规模图谱的工具。ODGI基于以节点为中心的对象(.og)运行,针对包含数百个单倍型解析基因组的泛基因组图谱进行了优化。比较注释工具包(CAT)也可以通过将参考基因注释投射到每个基因组上,对泛基因组图谱中的单倍型进行注释,这有助于简化物种内的注释工作。
3. 泛基因组可视化
泛基因组图谱可视化能够审视基因组之间的同源关系和变异情况,为潜在的生物学数据提供深刻见解。有多种工具可用于泛基因组图谱可视化。Bandage和GfaViz最初是为可视化组装图谱而创建的,它们可以生成二维图谱布局,允许以交互方式查看节点和边,变异在布局中以气泡形式呈现。vg viz 可以可视化节点、边、路径以及序列之间的碱基变异。SequenceTubeMap以一维 “管状图谱” 模型呈现这些元素,其中代表基因组的路径从左到右在图谱的序列节点间串联。
为了适用于千兆碱基规模的泛基因组,如人泛基因组,MoMI-G 将 SequenceTubeMap 的碱基层面可视化与Circos的染色体圈图相结合,以便高效浏览基因组之间的结构变异(SVs)和比对数据。ODGI 可以以二维或一维形式呈现图谱拓扑结构。Waragraph 是ODGI的交互式版本,目前正在开发中,旨在实现一维和二维可视化的查看。
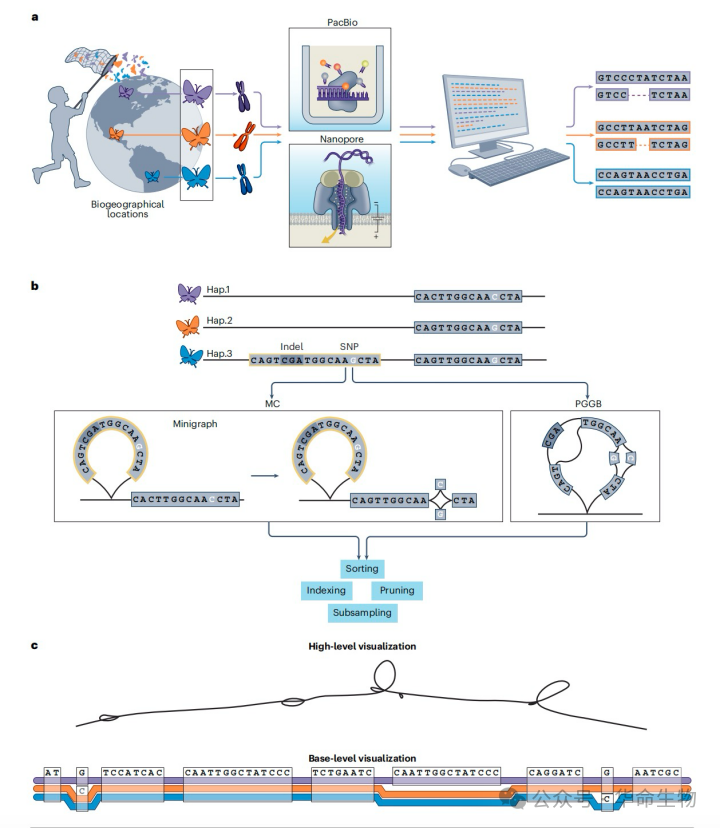
图2:泛基因组图谱构建流程
四、泛基因组下游分析与应用
1. 泛基因组展示变异
泛基因组图谱中的变异位点(单核苷酸多态性(SNPs)、插入-缺失(indels)和结构变异(SVs))可通过图谱分解来提取,图谱分解是将泛基因组图谱分解为更小、更易于处理的子图谱或元件(缠结或气泡)的过程 。图谱分解可使用vg snarl和gfatools bubble完成。在 MC和PGGB流程中实现的vg deconstruct,可以处理vg snarls的输出结果,或自动计算生成一个VCF文件。
结构变异可通过改变基因表达和塑造染色体重排等来影响适应性。完整呈现结构变异有助于分析基因组内的同线性和共线性。同时这可能为染色体进化提供见解,尤其是性染色体和微小染色体通常含有更多的结构变异,并且由于高重复序列和高GC而难以解析。人类泛基因组图谱能够识别异源近端着丝粒染色体之间的重组事件,尤其是在罗伯逊易位的断点处。这些易位是人类中最常见的染色体重排,而全面的泛基因组图谱极大地提高了对涉及SV的序列和形成机制的识别能力。
2. 群体基因组与转录组学数据分析
泛基因组图谱可在重测序项目中用作参考,以减少映射偏差。当参考图谱中呈现更多基因组序列,且嵌入已知变异时,短读长测序的映射可信度更高。由于标准算法无法直接应用于泛基因组图谱,需要开发新工具用于序列与图谱的比对。vg工具包中vg map适用于大型复杂变异图谱,vg giraffe目前正在扩展以支持长读长测序,它采用Burrows - Wheeler变换,支持从泛基因组图谱中高效查询和检索序列及变异。长读长测序也可使用GraphAligner进行比对。
提高短读长数据的比对率有助于重测序项目分析,尤其是在古DNA研究中。古DNA研究面临着污染、降解、内源性DNA量少、读长短等问题,因而比对率较低。将古DNA与变异图谱进行比对,与单个参考基因组相比,能够通过改善多态性位点的等位基因平衡来减轻参考偏差。
vg还支持将RNA-seq数据比对到可识别剪接的图谱上,生成的比对结果可用于量化单倍型特异性转录本的表达。泛转录组学有希望通过利用泛基因组参考图谱中嵌入的群体变异,高效量化单倍型特异性的差异基因表达。作者认为,将 RNA-seq 数据与泛基因组图谱参考相结合的泛转录组测序项目,将阐明基因流的影响,检测适应性遗传变异。染色质可及性分析,如ChIP-seq或ATAC-seq,也得益于泛基因组学,将它们与RNA-seq 数据相结合,提供了一种多组学方法,有助于解读对多种生物学过程和表型至关重要的调控事件。这些方法将推动未来在非模式生物中的泛表观基因组学研究,克服当前处理大型多组学数据集的局限。
3. 变异检测和基因分型
由于读长比对能力的提升,泛基因组图谱能够提高重测序研究中变异检测和基因分型的准确性。vg可用于从图谱中提取变异,并计算比对数据的覆盖度和比对质量,以准确识别已知变异。通过计算每个节点的数据覆盖度,可对较大的SVs进行基因分型。
PanGenie 中实现了一种用于已知变异基因分型的替代方法,速度更快,且无需数据比对。该算法结合了图谱中嵌入的长读长单倍型信息和短读长数据中的 k - mer,对未表征样本中的 SNP、indel 和 SV 进行联合基因分型。如果给定的变异区域被短读长k - mer覆盖较差,图谱中存在的单倍型可基于相邻变异来支持基因型推断。
在群体研究中,基于泛基因组的变异检测提高了准确性,并降低了每个样本的数据要求,有可能扩大可评估队列的规模。确定准确且全面的变异集可提高对种群历史、连锁不平衡和全基因组选择扫描分析的分辨率。这在有效种群规模较大的物种中非常有用,因为在这些物种中连锁不平衡程度较低。通过改进结构变异的基因分型,泛基因组图谱还有助于将结构变异整合到GWAS分析中,特别是现在越来越多的重测序项目采用三代长读长测序。因此,对基于泛基因组的SNP和结构变异数据集进行全基因组关联研究,能够增进研究人员对复杂多基因性状遗传基础的理解,揭示自然选择以及基因-环境相互作用和相关性的作用。
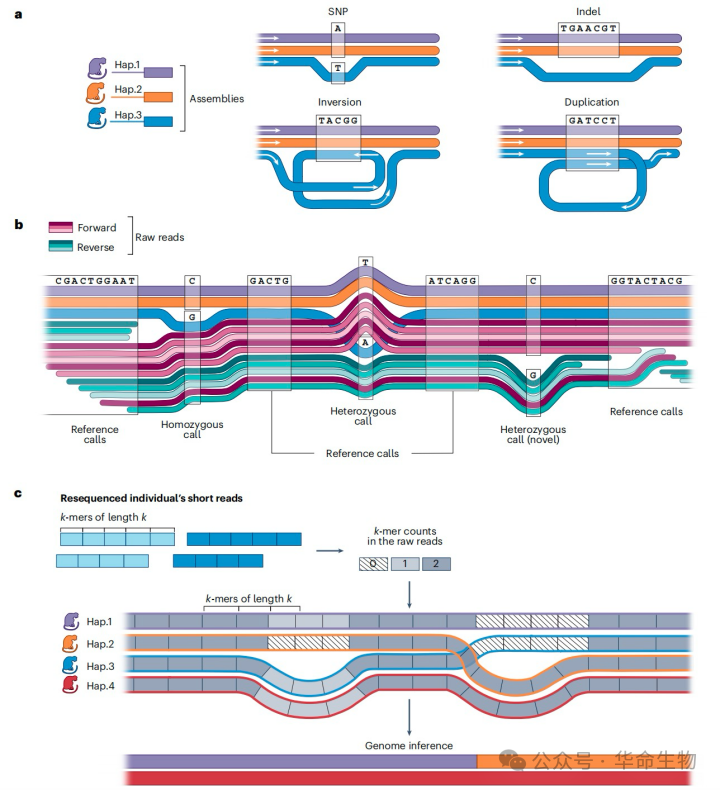
图3:泛基因组下游分析
结论与未来展望
在过去几年里,为生物多样性研究生成高质量参考基因组的大型项目计划如雨后春笋般涌现,包括脊椎动物基因组计划、达尔文生命之树计划以及欧洲参考基因组图谱计划等。地球生物基因组计划于2020年启动,提议在未来10年内对所有已命名的真核生物物种进行基因组测序。虽然目前仅有少数物种拥有泛基因组图谱,但泛基因组测序和组装方面的最新进展有可能将这一方法推广到大多数真核生物物种,有助于减少生物多样性、其演化及保护等所有相关分析中的代表性偏差。
由于成本和样本获取的限制,对许多物种而言,从多个个体收集、测序和组装泛基因组可能并不现实。在这种情况下,来自少数个体的泛基因组仍能提高代表性并减少参考偏差,尤其是对于高度杂合的种群,单个个体可能携带大量的等位基因多样性。泛基因组图谱可广泛应用于生物多样性的多个领域,从群体基因组学、系统发育基因组学、杂交与物种形成研究到保护基因组学,并且有望在未来成为此类研究的标准参考系统。目前,许多新方向正在探索之中。例如,泛线粒体基因组已被证明能改进个体的单倍型分析 ,并正被考虑用于从异质样本中进行物种鉴定。另一个有前景的新方向是超级泛基因组图谱,它将变异研究扩展到物种以上的分类等级,为研究物种分化、选择和重组过程背后的分子和进化机制,以及对快速气候变化的适应性开辟了新的可能性,特别是由于超级泛基因组图谱能够整合所有类型的基因组变异,它们有潜力阐明大型、随机交配且高度重组的野生种群复杂的进化历史和种系地理关系,以及改进诸如不完全谱系分选等事件的系统发育重建。超级泛基因组还可以协助研究发生杂交的复杂生态系统中的生物多样性。在泛基因组图谱中纳入两个杂交物种,将减轻因使用任一物种的参考基因组而产生的偏差,泛基因组还有助于揭示分化岛的起源,这些高度分化的基因组区域可能与生殖隔离相关,进而与物种形成过程相关。即使在同一物种内,一个包含所有亚种组装的全面泛基因组图谱,也能最大限度地识别特定亚种独有的结构变异。作者预测,随着未来用于比对不同物种泛基因组的工具的发展,物种水平的泛基因组也将在系统发育比较基因组学研究中取代线性基因组。
泛基因组图谱通过更全面地捕捉遗传多样性,也可能有效地指导旨在最大限度保护遗传变异的保护策略,特别值得关注的是与适应和应对环境压力相关的结构和功能基因组变异,这将改善在受威胁和濒危物种种群间重新引入和迁移个体的选择标准,更好地呈现结构元件,如结构变异、着丝粒和端粒、拷贝数变异以及单核苷酸多态性,连同非编码调控元件,可为近亲繁殖、远亲繁殖、有害突变、基因渗入和局部适应等提供全面的保护相关信息。泛基因组还可以帮助识别隐秘物种中的不同基因组区域,进而开发多基因座探针来区分隐秘分类群,简化保护管理工作。此外,作者设想泛基因组学能够通过提高古代DNA与近缘物种泛基因组的比对能力,帮助重建已灭绝生物多样性的基因组蓝图。对灭绝物种与其现存近亲进行更全面的比较,将有助于识别导致已丧失性状和生态系统功能的遗传变异,这些信息对于任何复活灭绝物种和恢复生态系统的努力都至关重要。
总之,随着组装、可视化、注释和分析泛基因组图谱的方法不断改进,作者建议生物多样性基因组学领域的研究人员都能接受泛基因组图谱这一研究新范式。
参考文献
"Pangenome graphs and their applications in biodiversity genomics“ Nature Genetics;https://doi.org/10.1038/s41588-024-02029-6
华命生物产品服务一览

华命生物目前已开通微信公众号、抖音、知乎、B站、小红书等线上平台,欢迎感兴趣的老师扫码关注了解更多内容!
