NG重磅 | 拟南芥泛基因组新图谱
拟南芥(Arabidopsis thaliana)为十字花科、拟南芥属一年生草本植物,因其基因组较小以及丰富的遗传多样性,成为了研究植物基因组演化和适应性的关键模型。然而,传统基因组学分析方法依赖于将短序列比对到单一参考基因组,这导致了对基因变异的理解偏向于简单、易对齐的区域,忽视了复杂的结构变异和转座元件的影响。
奥地利科学院的Magnus Nordborg及其科研团队于2025年8月19号在著名期刊《Nature Genetics》上发表了一篇名为“A comparison of 27 Arabidopsis thaliana genomes and the path toward an unbiased characterization of genetic polymorphism”的重要论文,通过分析27个拟南芥天然自交品系的基因组数据,系统揭示了结构变异与转座元件对基因组演化的深远影响,并指出短读测序和单一参考基因组在SNP检验中存在显著偏倚;该研究不仅为拟南芥遗传多态性提供了更全面的解析框架,也为其他物种的泛基因组学研究奠定了方法学基础。

一、基因组变异的组织
作者采用PacBio测序技术对拟南芥全球范围的27个样本进行基因组测序与组装,以揭示其基因组变异特征。结果显示,拟南芥基因组大小约为135-155 Mb,而实际可组装部分约120 Mb,差异主要来自着丝粒与rDNA等重复序列。长度长序列能够有效跨越转座子插入并顺利组装染色体臂,但在处理高重复的着丝粒和45S rDNA簇时仍存在困难。尽管不同转座子家族在各采样点间差异显著,但其对整体基因组大小的贡献有限,远小于其在物种间变异中的作用。研究表明,拟南芥基因组大小变异主要是由于着丝粒和rDNA重复序列所造成,而非转座子累积效应。
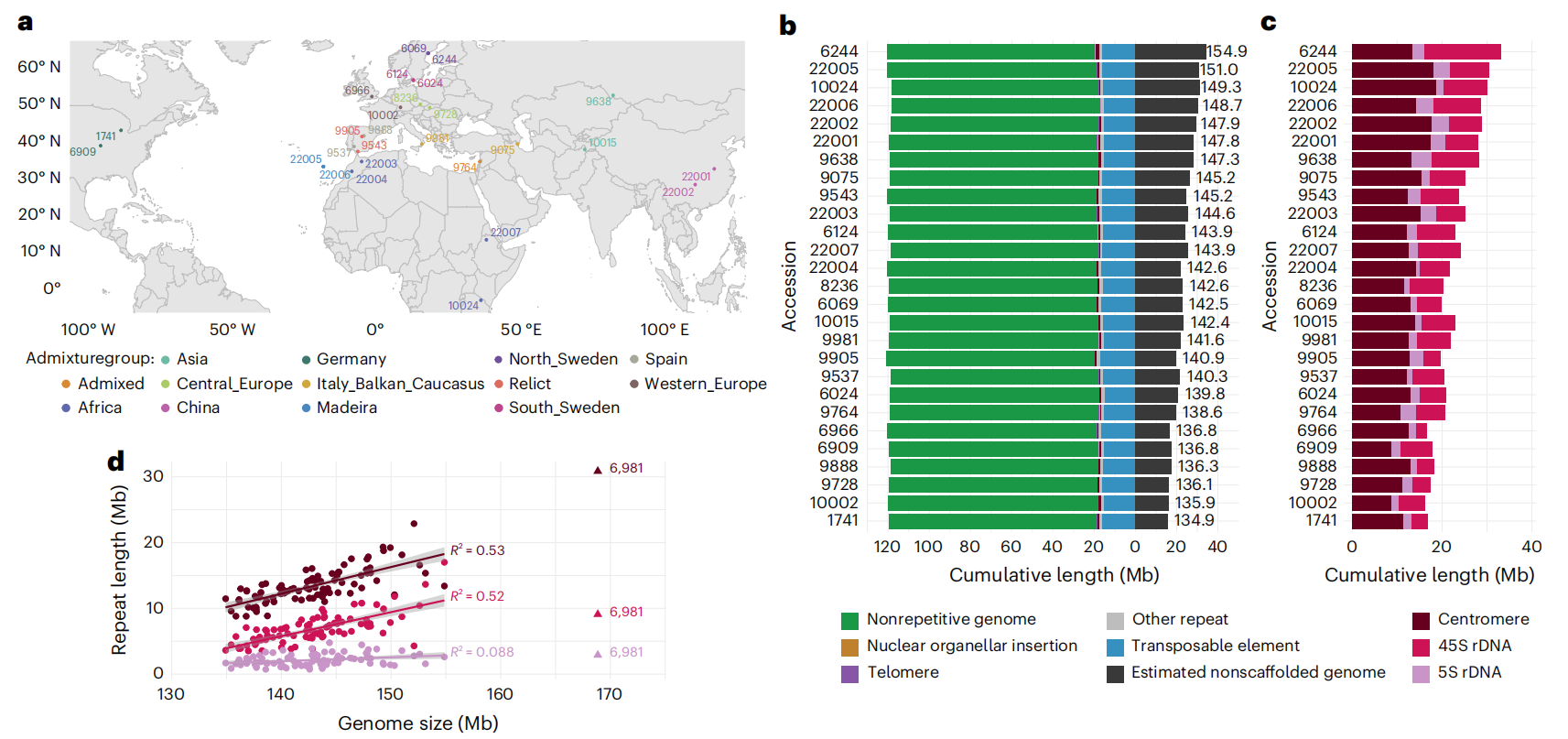
图1:27个拟南芥种质的基因组组装和大小变异
二、结构变异检测
作者发现,不同来源的拟南芥其染色体臂在长度和基因顺序上整体保持一致,但仍检测到13Mb的结构重排事件,例如第4号染色体上的1.2 Mb倒位,以及长江流域样本“22001”中的大范围相互易位。作者指出,与单核苷酸多态性(SNP)相比,结构变异(SV)的检测更具挑战性,因为结果往往依赖比对方法和参数设定,目前还没有统一的标准。为了解决这一问题,作者采用了两种互补的方法:全基因组多重比对流程Pannagram和泛基因组图构建工具PGGB。作者发现,PGGB的检测范围更广,能够覆盖Pannagram的结果,而Pannagram则提供了更直观、易于解释的变异信息和统一的坐标体系。因此,作者在后续分析中主要基于Pannagram展开。
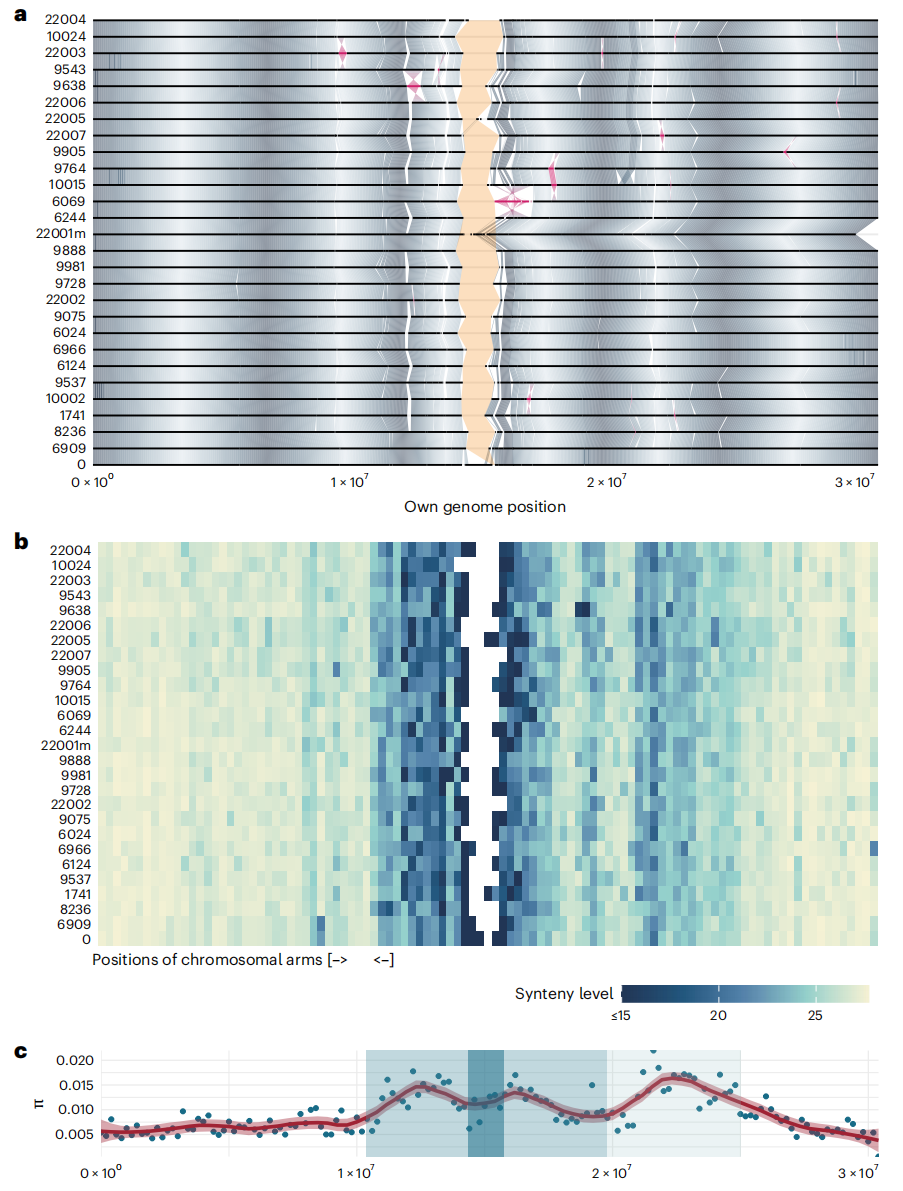
图2:1号染色体变异的分布
三、SVs的特征
作者重点分析了数量最多的长度变异,并将其分为简单型sSV和复杂型cSV,其中sSV占比超过80%。研究共识别出53万余个sSV,覆盖37.5Mb的基因组区域,且长度分布偏向短变异。进一步分析发现,大多数sSV源于稀有的长插入和短缺失,并主要发生在基因间区,在基因区内则更常见于内含子。此外,作者还发现了108个来源于细胞器的插入,规模从数百bp到完整基因组不等。这些结果表明,sSV不仅在数量上占主导,还受选择压力约束,并揭示了细胞器DNA持续转移到核基因组的现象。
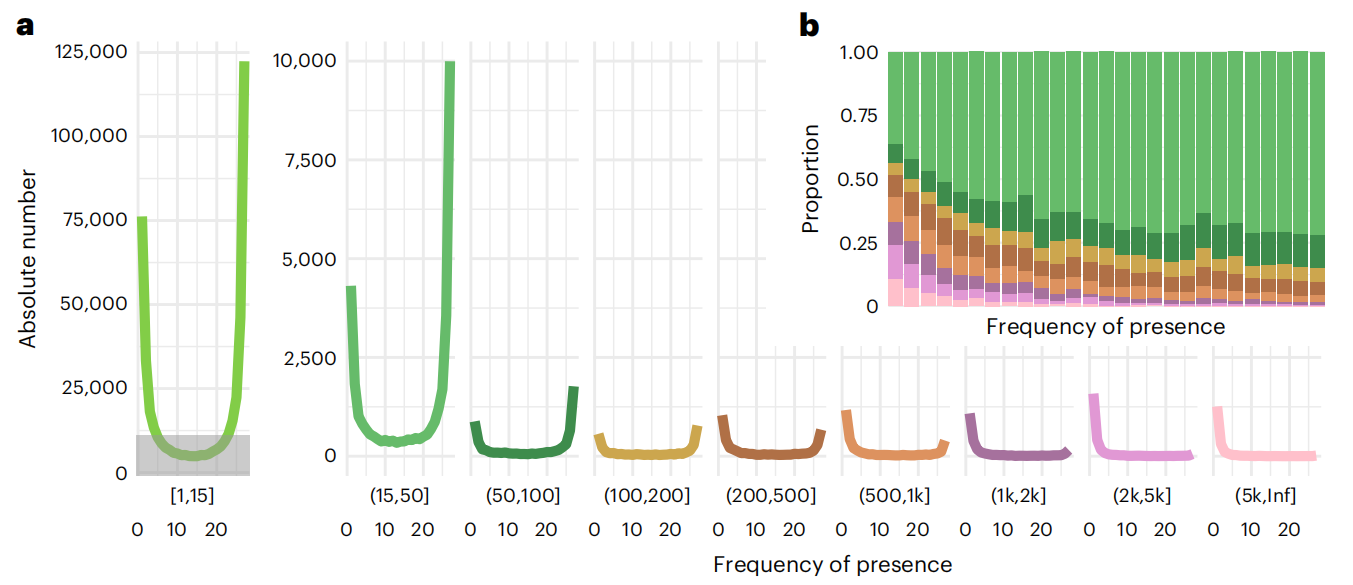
图3:SVs等位基因频率分布
四、SVs和注释TE
作者进一步分析了SV与转座子(TE)的联系,结果显示超过 60% 的sSV与TE注释重叠,说明转座子活动是导致结构变异的重要来源。作者发现,可能的插入通常比缺失更长,且往往对应完整的TE,提示其与近期的转座子活动相关;而可能的缺失多对应残缺的TE,表明这些序列正在衰退。值得注意的是,完整的TE相关sSV在长度上呈现特定富集(约5 kb),与活跃转座子的特征一致。类似的富集模式在其他类别的sSV中也有体现,除了与TE片段相关的部分。这一结果支持了近期观点:目前对活跃TE的注释仍不完善。
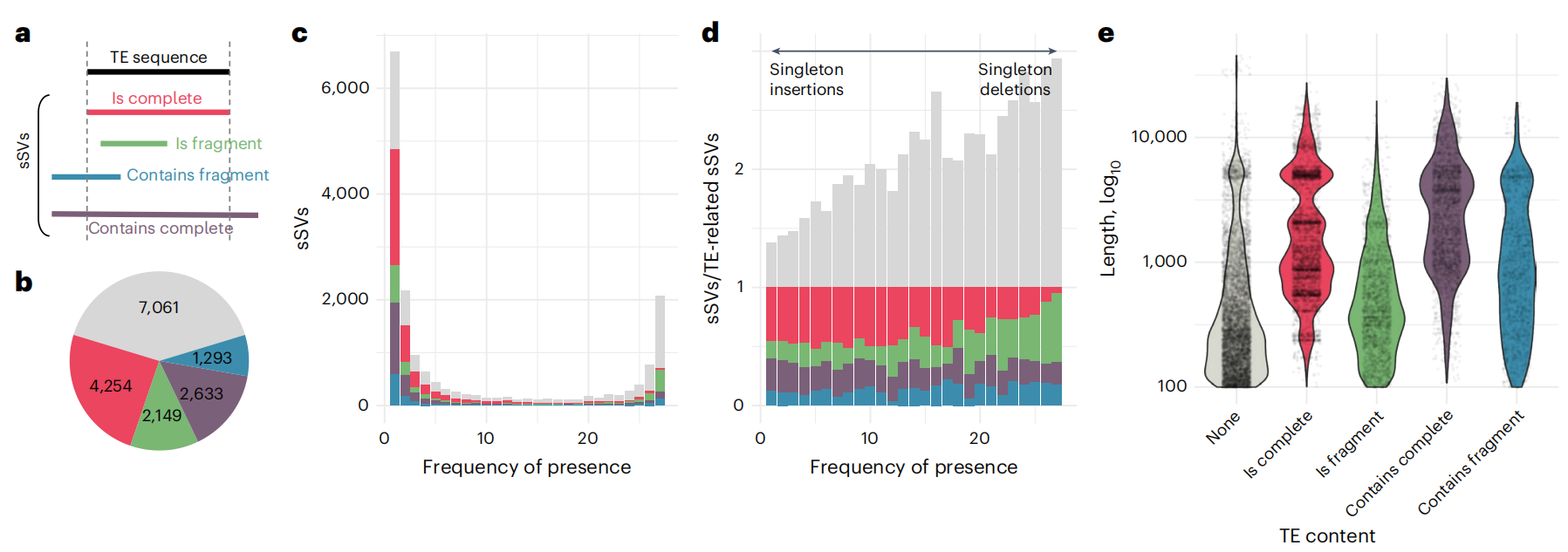
图4:TE在双等位基因中的作用
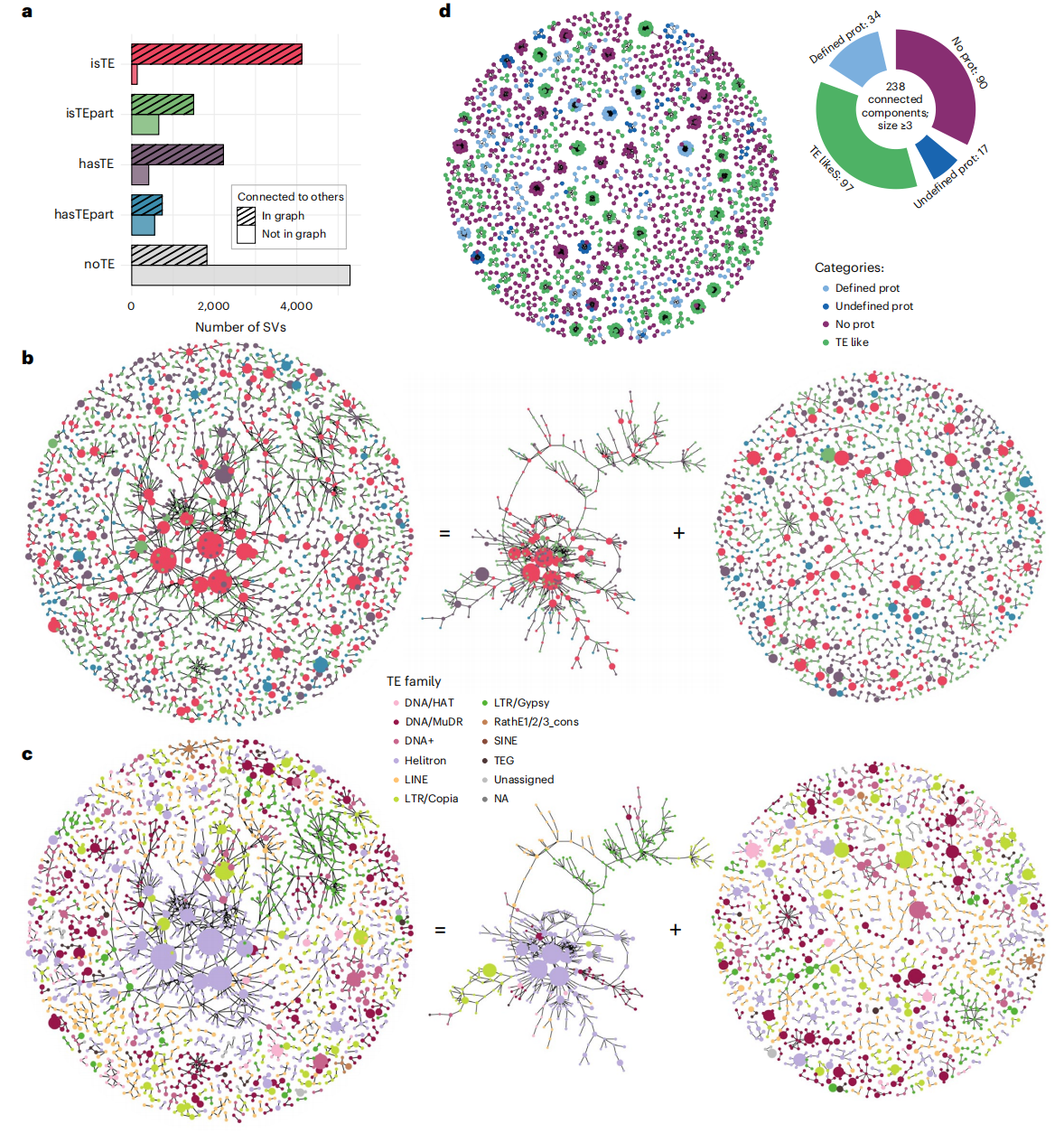
图5:sSV结构图
五、移动元件组mobile-ome研究
作者在文中首次提出“移动元件组”(mobile-ome)的概念,用以系统刻画基因组中全部可移动遗传元件及其对基因组结构与变异的贡献。通过改进的全基因组比对与序列相似性分析,作者发现大量此前未注释的转座子及其片段,表明现有注释体系远未完整。研究进一步揭示,这些移动元件不仅分布广泛,还表现出不同的活性状态:完整元件多与近期的插入事件相关,而不完整或退化片段则反映其衰退过程。作者指出,移动元件通过插入、缺失及介导DNA修复等机制推动结构变异的形成,并在染色体臂和非编码区中尤为活跃。同时,它们往往与表观遗传沉默机制相关,显示出宿主在限制其扩增方面的作用。
六、基因组集gene-ome研究
作者又进一步提出“基因组集(gene-ome)”的概念,用以研究所有蛋白编码基因的整体特征,并采用独立注释与RNA测序结合的方法,尽量减少参考基因组偏倚。结果共注释到34153个基因,其中约2661个PC基因和565个TE基因此前未被记录。分析27个材料发现,约13%的基因呈分离状态,这些变异主要源于新基因插入,而非祖先基因缺失。进一步比较显示,分离基因更常见于着丝粒附近,而固定及祖先基因更多分布在染色体臂区,说明核心与边缘区域在进化中承担不同角色。功能上,新基因富集于防御相关通路,而旧基因偏向管家功能。表达上,固定基因活跃度更高,而分离基因和非祖源基因往往低表达并更易受到表观遗传沉默。这一分析揭示了拟南芥基因组在保持稳定核心的同时,不断通过基因新增与转座子活动维持多样性。
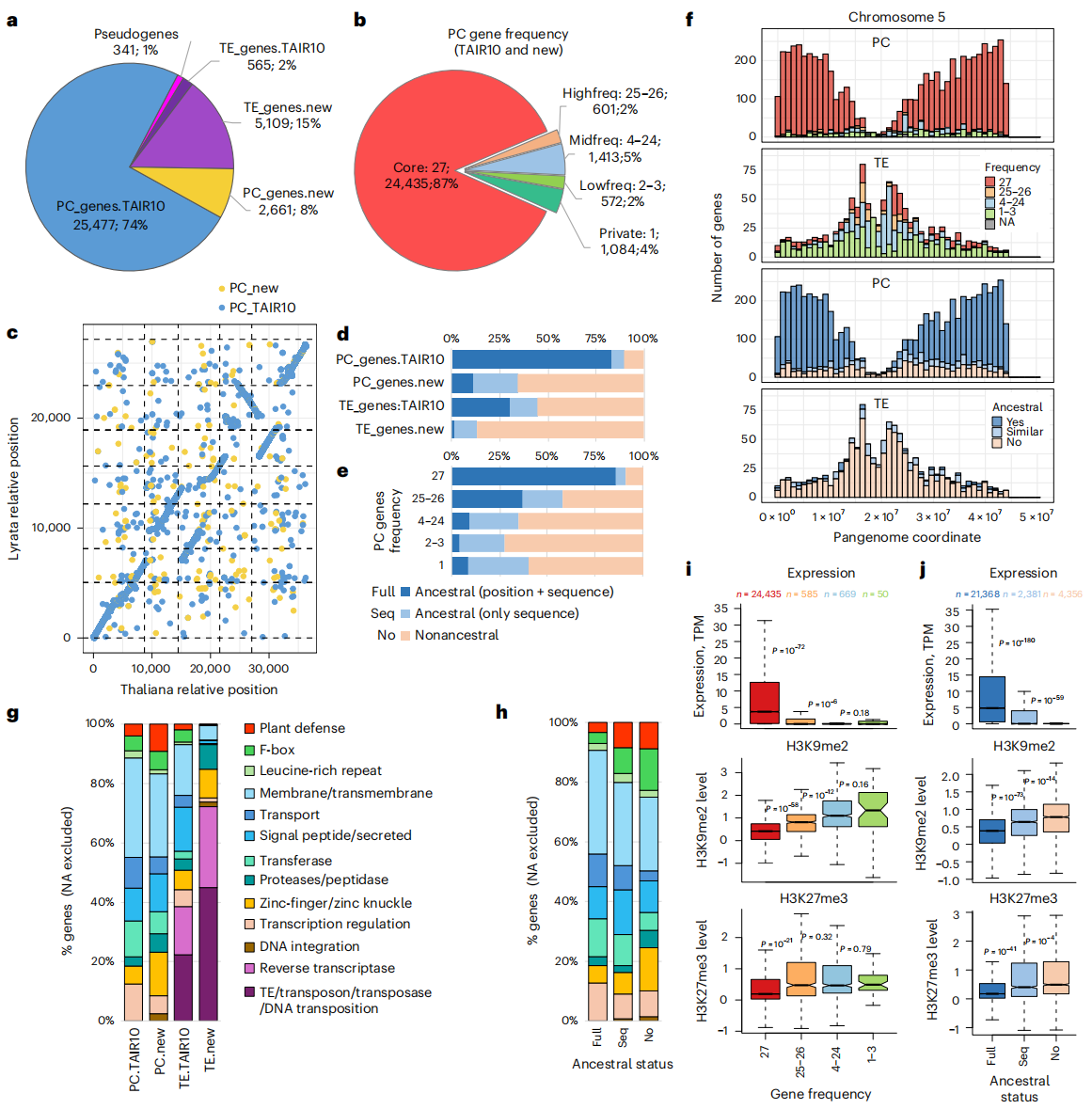
图6:基因组集(gene-ome)的分析
七、泛基因组研究
在拟南芥的研究中,作者发现泛基因组随样本数量增加而呈现差异化增长趋势:可移动元件的扩增速度快于完整基因组,而基因部分因受更强的纯化选择而增长较慢。作者进一步推测,整体增长速度高于恒定种群中性进化下的对数模式,但低于指数扩张种群中的线性预期;同时,变异的空间分布并不均衡,主要集中在着丝粒区域。研究结果显示,仅在27个拟南芥群体样本条件下,泛基因组染色体长度较TAIR10参考基因组延长高达63-76%。
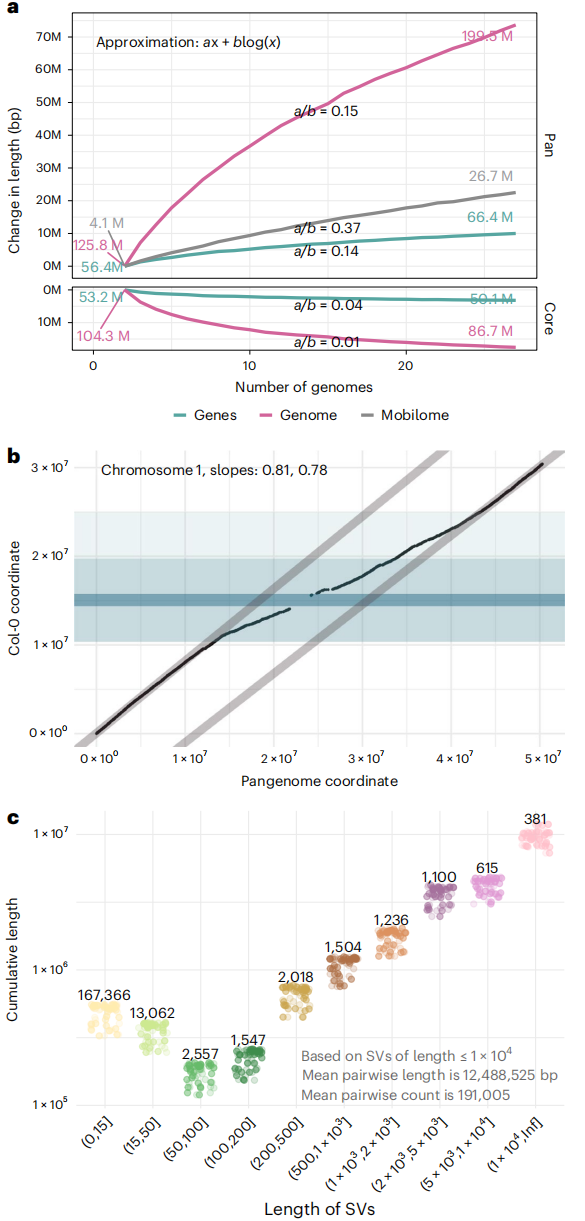
图7:泛基因组的增长
八、缺失的多态性
作者在本研究中基于拟南芥数据系统评估了以往重测序数据中遗漏的多态性。此前的“1001基因组计划”利用短读段仅检测到部分SNP和短SV,平均覆盖率约为84%,导致25-45%的SNP变异未被识别。通过全基因组比对,作者发现两份基因组平均存在约19万个SV,覆盖约12.5 Mb序列,相当于每个基因组约10%的长度。进一步分析表明,短读段漏检SNP的主要原因是比对区域覆盖不足及参数设定差异;采用PCR-free高覆盖度数据后,缺失率可降至20%以下,但FDR仍接近7%。在分析过程中,参考基因组中缺失的分离重复(segregating duplications)会导致大量的伪杂合性(pseudoheterozygosity)信号。
九、参考基因组偏倚
作者进一步在系统评估了参考基因组的选择对分析结果的偏倚影响。作者认为,SNP检测的错误率不仅取决于参考基因组本身,还受到参考基因组与样本基因组亲缘关系的影响。因而这种参考偏倚对依赖SNP进行群体遗传学推断的研究具有潜在风险,可能影响全基因组关联分析和群体历史推断等下游应用。进一步比较RNA-seq和甲基化组分析(BS-seq)数据比对到TAIR10参考基因组与样本自身基因组的结果表明,两者表达量大部分高度相关,但仍有部分基因差异显著,主要富集于拷贝数可变基因(CNV genes)。同时,BS-seq对参考基因组的选择更为敏感,当把数据比对到参考基因组而非样本自身基因组时,会产生大量假阳性的差异甲基化区域。作者指出,参考基因组带来的偏倚问题在不同研究中影响程度不同,但必须予以重视,并在分析和解读结果时谨慎考虑。
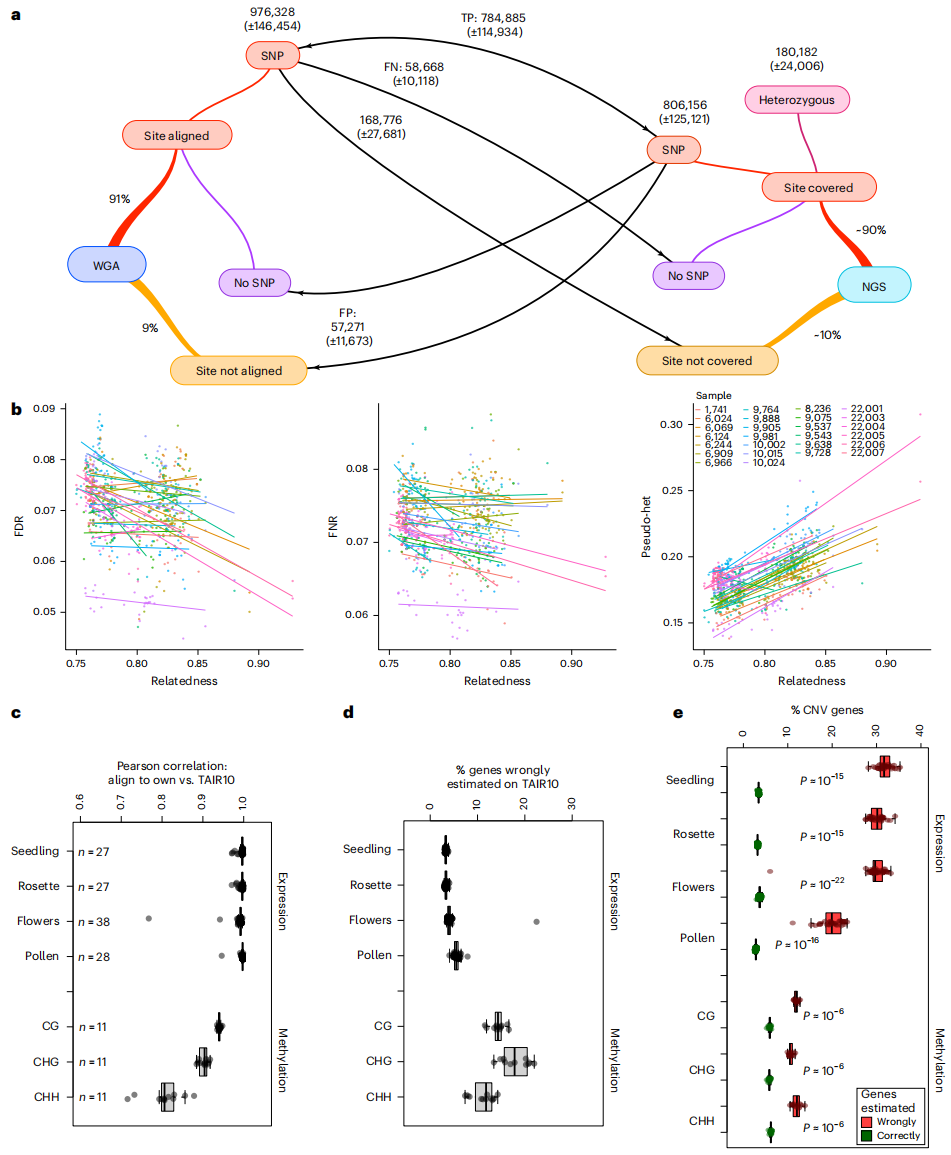
图8:读取映射和参考基因组偏倚
结语
本研究突破了传统依赖短读长测序和单一参考基因组的局限,基于27个拟南芥自然自交品系的全基因组数据进行了系统分析。结果显示,不同品系的基因组大小差异主要源自着丝粒区和rDNA卫星重复的变异;而染色体臂虽在长度和结构上保持高度保守,却存在大量由转座元件(TEs)插入驱动的结构变异。通过构建“泛基因组”坐标体系,作者发现其规模随样本量显著扩展,在样本数为27时已比任何单一基因组大约70%。在基因层面,约13%的基因表现出品系特异性缺失或新增,多数为非祖先来源,且常受TE样表观遗传沉默;而祖先基因则保持更高表达。进一步分析揭示,短读长测序在SNP检测、转录组及甲基化分析中存在明显参考偏倚,凸显多基因组参考体系的必要性。本研究不仅首次实现了拟南芥遗传多态性的全面评估,也为大基因组和高多态性物种的群体基因组学研究提供了重要方法学参考,具有深远意义。
华命生物产品服务一览
华命生物目前已开通微信公众号、抖音、知乎、B站、小红书等线上平台,欢迎感兴趣的老师扫码关注了解更多内容!