Cell Genomics综述 | 泛基因组解析基因组进化动态
随着基因组学研究的深入,传统的线性参考基因组已无法全面揭示物种间和群体内的基因组多样性。尤其是在结构变异和附加基因(如存在缺失变异和非参考序列)方面,单一参考基因组的局限性愈加显现。为了解决这一问题,泛基因组应运而生,作为一种新的基因组参考体系,它整合了多个个体的基因组序列,能够捕捉更为全面的基因组多样性。
国外研究团队于2025年11月18日在国际著名期刊Cell Genomics上发表了一篇标题为“Dynamics of genome evolution in the era of pangenome analysis”的综述性论文,重点探讨了泛基因组的核心优势与应用:通过整合多基因组数据来更全面地捕捉结构变异,揭示遗传多样性与表型变异之间的联系,并进一步深入分析了其在基因型-表型关联研究中的潜力及挑战。

一、现代视角下的泛基因组构建与完整性
泛基因组的概念自 2005 年提出后不断发展,从最初的基因集合扩展到描述群体整体遗传多样性的参考框架。与依赖单一基因组的线性参考不同,泛基因组整合多个个体的数据,以更全面地呈现变异与结构差异。为达到遗传多样性“饱和”,需要根据物种的多样性选择适当数量的样本:低多样性物种只需几十个,高多样性物种如玉米与酵母则需数百个,以达到约 90%-95% 的基因含量覆盖。除样本数外,基因组质量也是完整性的关键影响因素。映射策略成本较低,但难以捕获复杂结构变异;组装策略分辨率更高,但质量不一、连续性不足会导致重排缺失。人类泛基因组项目清晰呈现了泛基因组构建的主要挑战:在不断扩大样本规模的同时保持组装质量。从 T2T-CHM13 的问世到大量单倍型组装的建立,这一过程中始终存在取舍。随着测序与分析技术的提升,这些限制在未来有望被逐步克服。
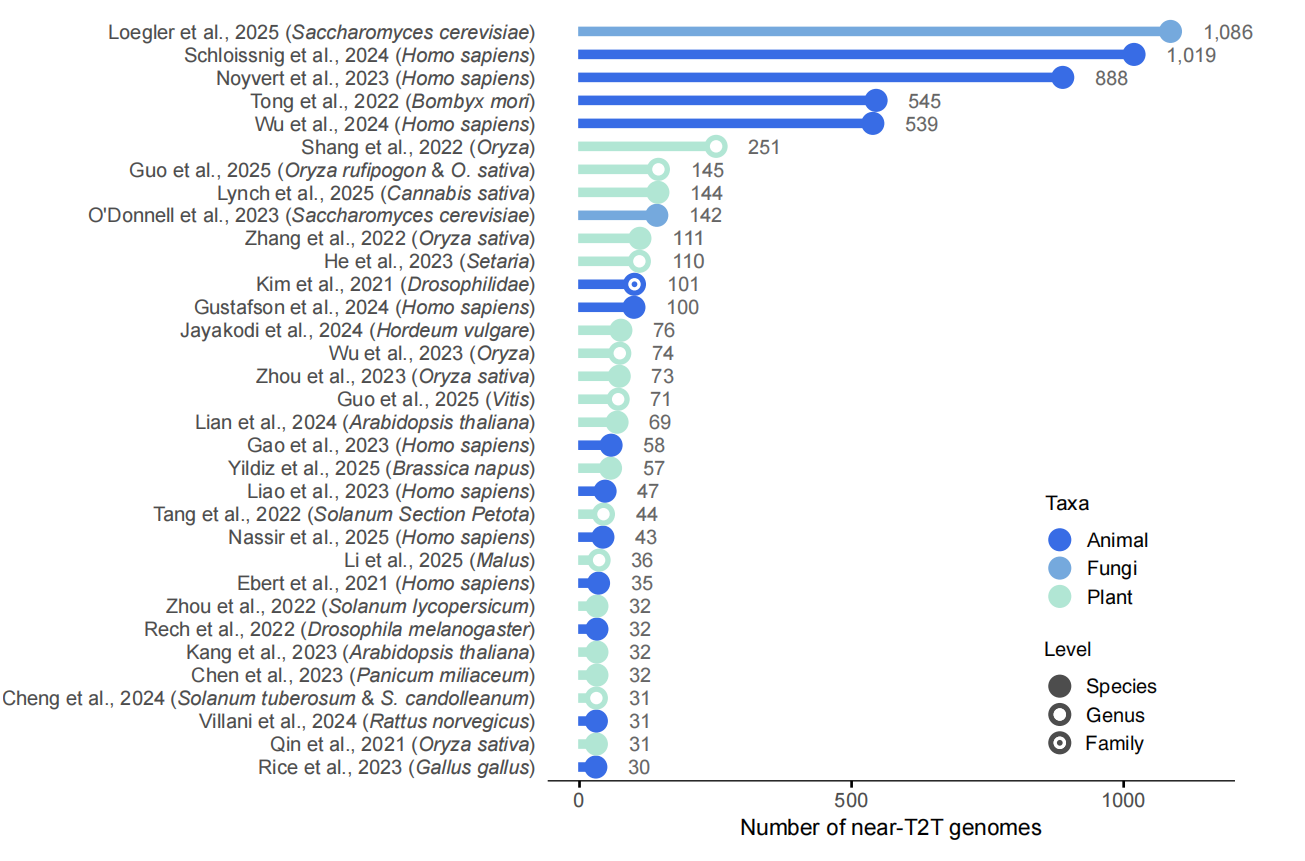
图1:泛基因组研究包含的基因组数
二、通过图结构泛基因组捕获NRSs
图结构泛基因组的优势在于能捕获线性参考中缺失的新序列(NRSs),从而更全面反映群体遗传多样性。NRS的多少既取决于群体本身的基因多样性,也受构图方法影响。当前主要有两类策略:一类基于长读长比对或组装比较检测结构变异,并将这些变异整合入图结构,可在多种物种中显著增加参考序列长度,如水稻、番茄、玉米等物;另一类依赖全基因组比对整合同源区域,计算量更高,但能捕获更复杂的变异,在牛、拟南芥和葡萄属等群体中带来更大范围的参考扩增。泛基因组还可进一步简化为非冗余序列,以更准确量化真实新增的序列,在大麦和酵母中观察到约27%–33%的非冗余增加。人类泛基因组研究显示,NRS的检测量随样本质量和算法不同而变化:47个高质量样本能检测175-190Mb的NRS,而539个质量较低的样本约检测60Mb;区域性泛基因组(如中国、阿拉伯和太平洋人群)进一步捕获族群特异的新序列,并提升比对准确性与变异检测敏感度。
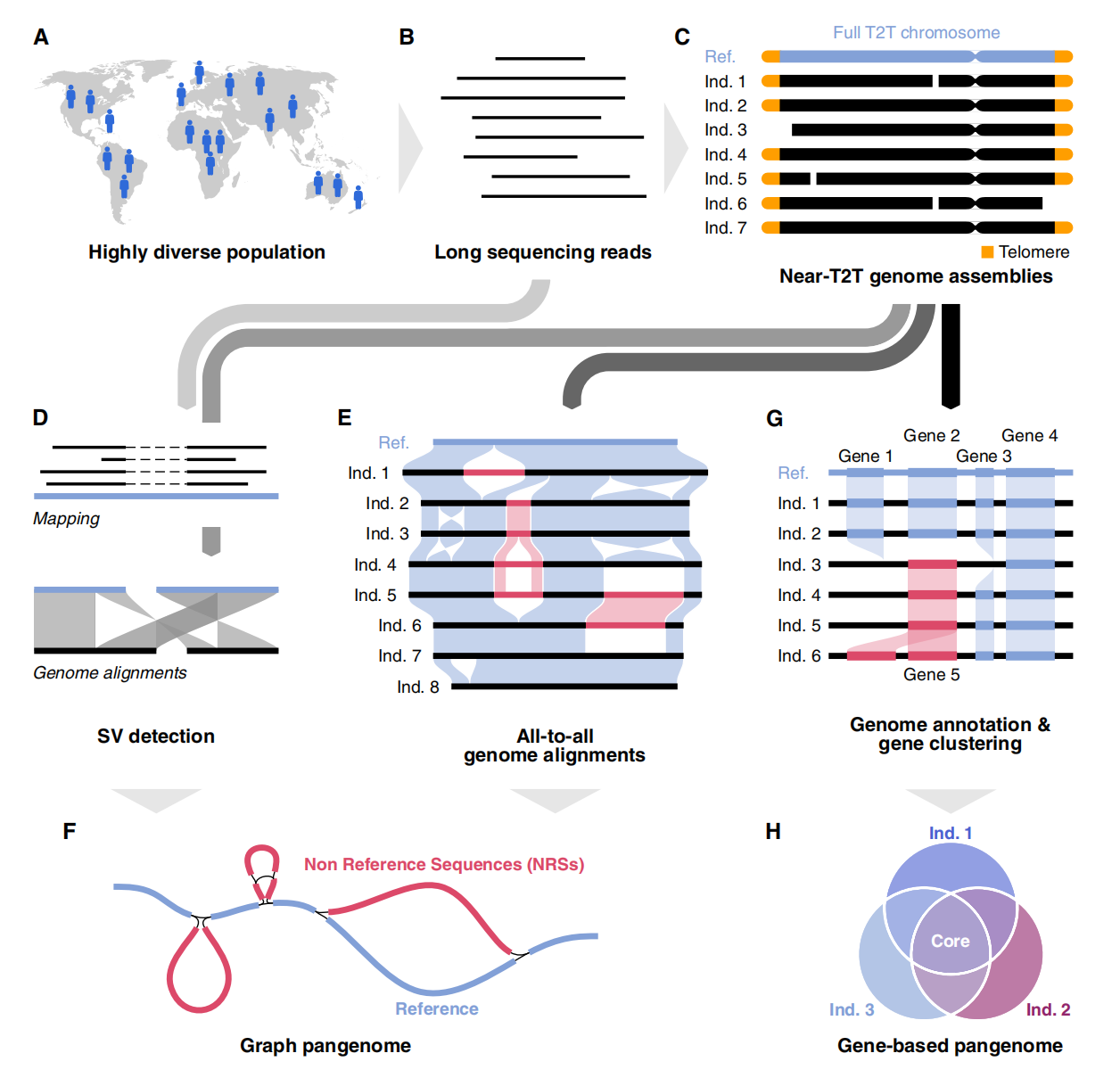
图2:泛基因组的构建
三、通过泛基因组分析获得的基因流和基因渗透见解
基因流机制,包括古代杂交事件和水平基因转移(HGTs)引起的基因渗透,在泛基因组中起着重要作用,推动了NRSs的形成。使用near-T2T基因组的单倍型分型技术,能够精确界定基因渗透区块的边界和序列。例如,中国泛基因组的研究通过表征704 Mb的渗透序列,揭示了东亚基因组中丹尼索瓦类古老基因渗透的富集;在酵母中,T2T基因组的组装帮助揭示了染色体尺度的基因渗透结构,并精确表征了对端粒多样性贡献较大的HGTs。
基因流的后果之一是基因位点的系统发育与基因组广泛系统发育之间的偏差,这可能是由于不完全谱系分离(ILS)导致的。为了避免ILS偏倚,可以使用ABBA-BABA检验,这一方法在泛基因组分析中得到了广泛应用。尽管这一方法已经产生了有价值的结果,特别是一些渗透基因对抗逆性的贡献,但SNP计算方法仍未完全发挥泛基因组数据的优势。此外,基因流还会通过减少单倍型分化影响群体间遗传多样性。通过全基因组比对,该方法揭示了水稻、苹果等作物中抗性基因的渗透,表明基因渗透在作物驯化过程中具有重要作用。尤其在水稻中,发现了一个重叠着着丝粒的4.48 Mb渗透区块,验证了基因流在遗传资源流动中的重要性。尽管一些方法在识别低频变异时面临挑战,但泛基因组数据的应用,为跨群体基因流和渗透现象的研究提供了强有力的支持。
四、真核生物泛基因组中结构变异多样性的分布格局与成因
在泛基因组分析中,除了揭示新序列外,结构变异(SVs)的识别同样至关重要。SVs通常分为四类:PAV、CNV、易位和倒位。借助长读长测序和近T2T基因组,使用比对和图结构解构等方法能够显著提高SV的检测精度。SV的形成与基因组结构紧密相关,转座元件(TEs)在其中起到了重要作用,尤其是在多倍体物种中,TE相关的SV占总SV的23%至86%。此外,SV常富集于重复序列和片段复制区域,显示了基因组架构对SV形成的深远影响。SV在基因组中的分布不均匀,多项研究揭示了SV“热点”区域,如染色体末端、rDNA区和抗性基因附近。在不同物种中,SV的多样性差异显著,植物通常比动物表现出更高的结构变异,这可能与全基因组复制(WGD)有关,WGD增加了突变输入并减少了纯化选择。随着倍性水平升高,SV数量也会相应增多,所以许多植物作为古多倍体保留了较高的结构变异。在核苷酸层面,植物通常比脊索动物表现出更高的多样性。节肢动物则表现出较高的核苷酸多样性,某些物种(如家蚕)也表现出较高的SV多样性。综上,跨群体SV研究为基因流、物种适应与演化提供了深刻的遗传学见解,且随着样本规模的扩大,泛基因组数据的解析能力将持续提高。
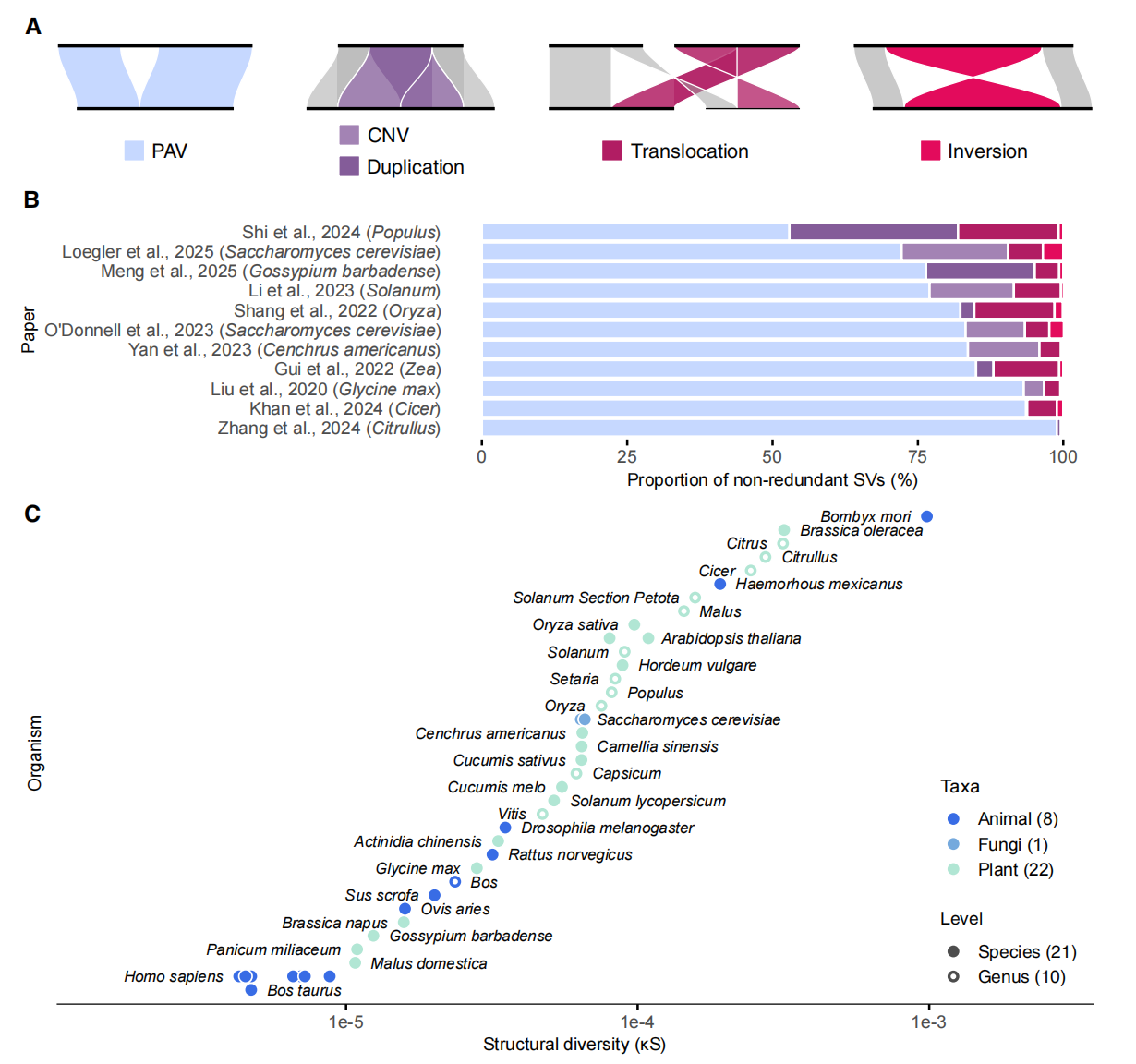
图3:结构变异的多样性
五、基于基因的泛基因组的功能性和进化性见解
基于基因的泛基因组不同于以完整基因组序列为基础的图结构泛基因组:它从功能层面出发,整合物种或属内所有个体的基因集合。具体而言,该方法首先对高质量基因组进行统一注释,然后依据序列相似性与/或基因共线性将预测基因聚类为正交组。随着群体内个体遗传分化程度加深,正交关系的识别难度显著上升,因此有必要针对不同群体动态调整相似性阈值。近T2T基因组提供了更完整的基因内容与更可靠的共线性信息,从而显著提升了基于基因的泛基因组构建精度,并已在水稻等作物研究中得到验证和应用。
依据基因家族在群体中的出现频率进行分类:核心基因在所有个体中共享,而辅助基因则在出现频率上表现出较大差异,并进一步划分为软核心、可有可无和私人基因。基因频率分布与泛基因组的开放性密切相关;真核生物往往具有较高比例的核心与软核心基因(60%-88%),表现出接近封闭的泛基因组结构。大量比较研究揭示了核心与辅助基因在功能上的显著分化:核心基因富集于基本与必需的生物过程,而辅助基因更多参与次级代谢、环境适应与应激响应,并在物种生态适应和驯化过程中发挥重要作用。例如,野生种特有的辅助基因与抗逆性密切相关,而驯化群体则更富含与发芽、营养或品质性状相关的基因。在表达层面,核心基因通常具有更高表达量并覆盖更广泛的组织范围,进一步反映了它们在生物体生存和稳态维持中的关键地位。
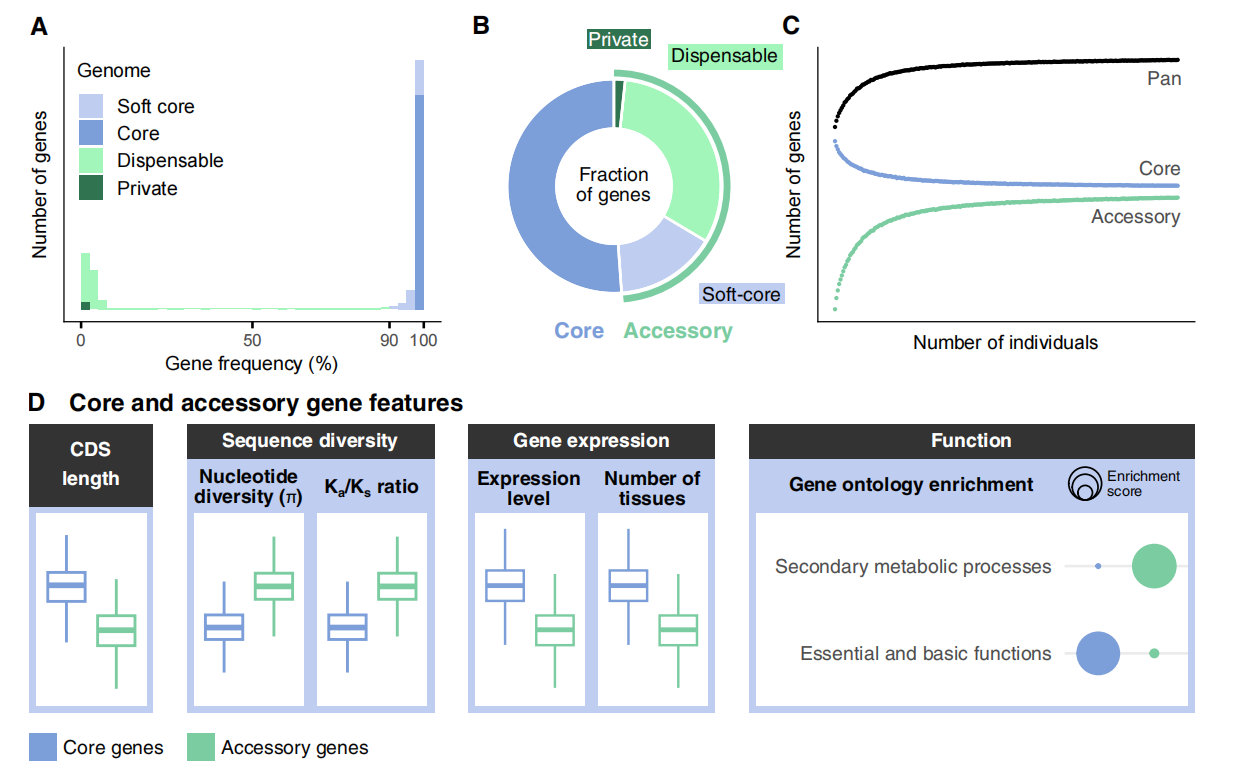
图4:基于基因的泛基因组及其相关特征
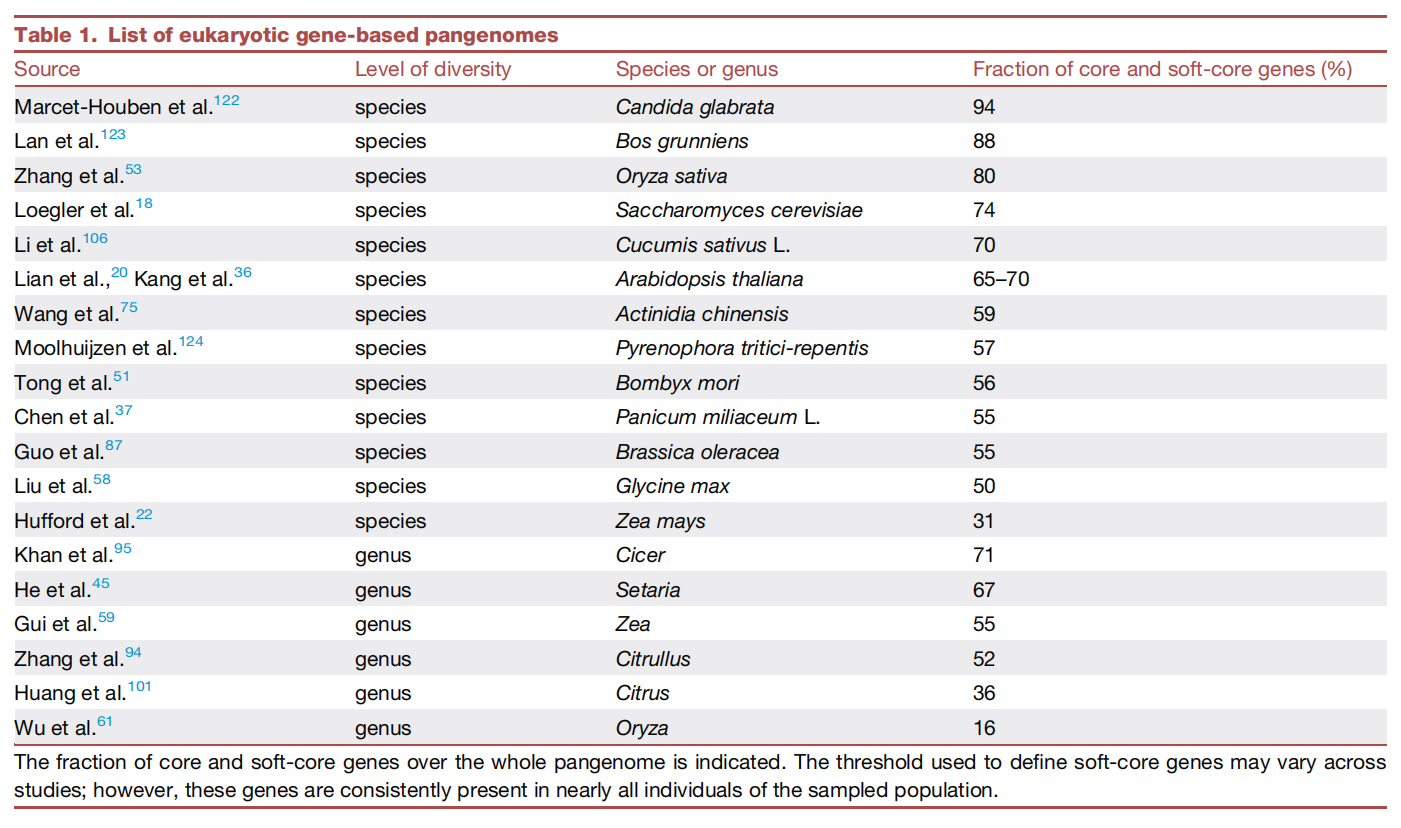
表1:真核生物基因泛基因组列表
六、利用泛基因组推动 SV–性状关联的发现
结构变异可显著影响生物表型,已在多物种中与疾病抗性、适应性和形态变化等性状建立关联。然而,要全面评估SV对群体表型变异的整体贡献仍具挑战性,因为SV相关的GWAS和QTL分析需要大规模且匹配的基因型—表型数据。泛基因组为此提供了高效框架,通过近T2T基因组构建的代表性图结构即可捕获常见SV,无需为所有样本生成高质量组装,并可利用短读长在更多样本中进行低成本SV分型,从而支持大规模SV–性状关联研究。
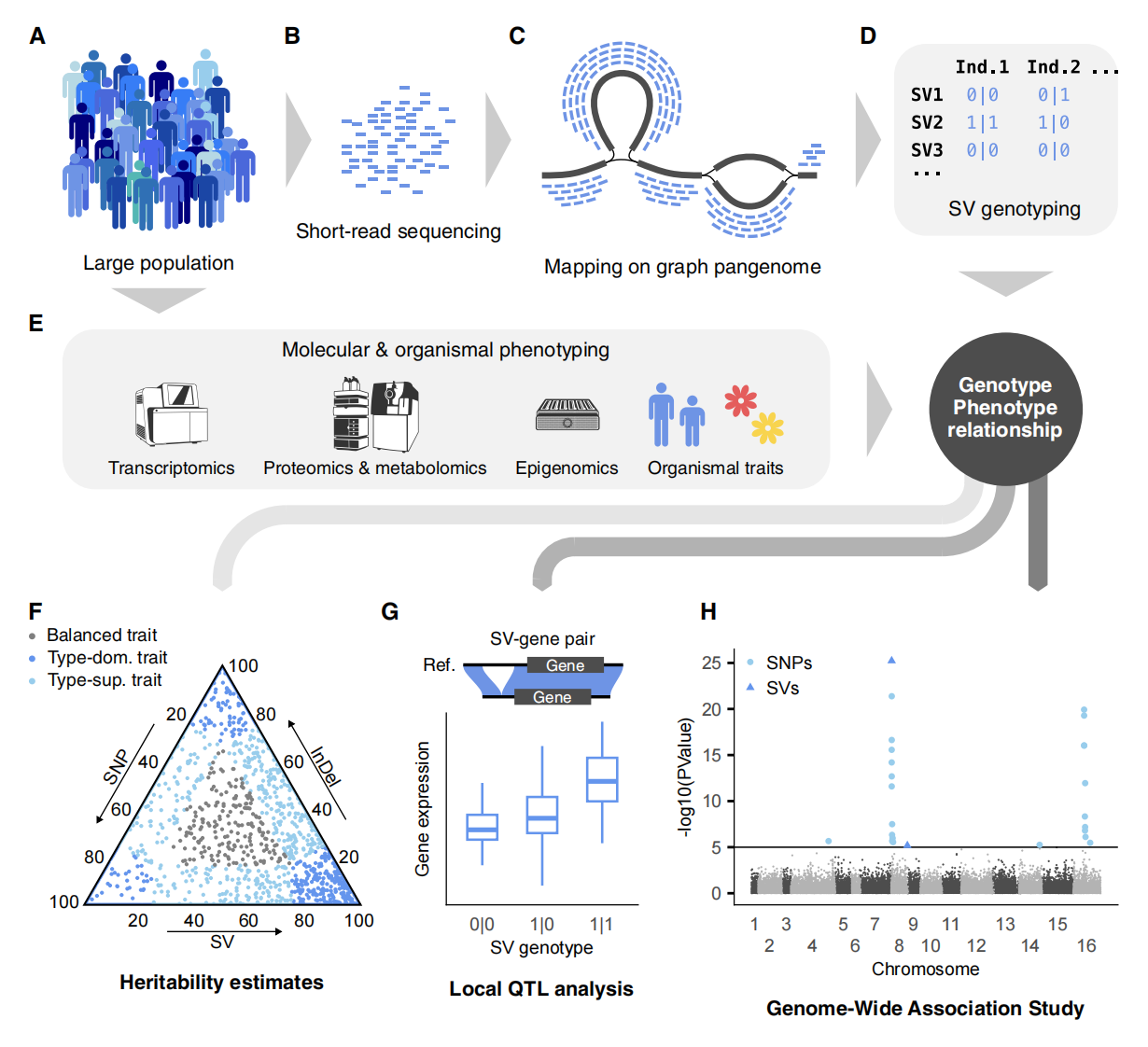
图5:基于泛基因组的基因型-表型分析
七、SVs 在塑造分子性状中的作用
SV在分子性状的形成中具有关键作用,数量性状从可见表型延伸至基因表达、剪接、代谢物与表观遗传标记等多层级分子特征。泛基因组的应用使得解析SV对这些性状变异的贡献成为可能,尤其在基于图结构的基因分型体系中,可以系统评估SNP、indel与SV的相对影响。多项研究显示,在番茄、玉米等作物中,将SV纳入模型显著提高了分子性状遗传力的解释度,且相当比例的性状变异呈现SV主导。局部SV常导致基因表达差异,但显著的SV–基因关联仍相对有限。尽管SV相关的GWAS受限于样本规模与表型数据获取,其在生长及复杂性状中的重要性已得到反复验证。SV可通过编码区与调控区两种路径影响分子性状:CNV与PAV通过剂量效应改变表达水平,而易位与倒位则通过改变基因的调控环境产生广泛影响。这些结果共同强调,SV是驱动分子性状多样性的核心遗传因素。
八、SV 与生物性状变异的关系
SV 在生物性状变异中发挥重要作用,尤其在农艺性状的研究中。改良基因和相关的 SV 在栽培品种中通常比地方品种更为丰富,并且在强选择压力下,这些变异位点往往表现得更加明显,因此成为重要的候选位点。例如,在蚕中,多个与改良相关的 SV 和基因与丝绸产量变异相关,功能验证显示敲除该基因可显著减少丝绸产量,而过表达则能提高产量。此外,基于 SV 的 GWAS 已被广泛应用于多个农艺性状的研究,如稻米产量、牛头色和西瓜甜度等。经济相关的 SV 在酵母中也得到了发现,例如 CUP7 基因的重复与抗铜硫酸盐能力相关,而易位与 SSU1 基因的表达增加则增强了抗亚硫酸盐的能力。图结构泛基因组使得 SV 可以与现有的 GWAS 数据集结合,帮助重新分析农艺性状。然而,由于大多数研究集中于有限性状,目前还无法得出 SV 对生物性状变异的普遍结论。
总结与展望
长读长测序的应用推动了T2T泛基因组的构建,使得从SNP到大规模SV的遗传变异能够全面检测。这些泛基因组不仅深化了对SV分布、核心基因与辅助基因特征的理解,还促进了基因型-表型之间的关联研究。尽管大规模表型分析仍面临挑战,早期研究已揭示SV在表型变异中的重要作用。构建全面的变异目录不仅依赖广泛的变异检测,还需要覆盖多样的基因组样本,尤其是非人类动物物种。目前,泛基因组的可扩展性仍存在困难,现有工具和格式更适用于线性参考,而图结构表示尚未得到充分发展。因此,发展新的数据格式,解决群体变异的全面检测,成为突破现有限制的关键。
华命生物产品服务一览
华命生物目前已开通微信公众号、抖音、知乎、B站、小红书等线上平台,欢迎感兴趣的老师扫码关注了解更多内容!